Downloaded 105 times






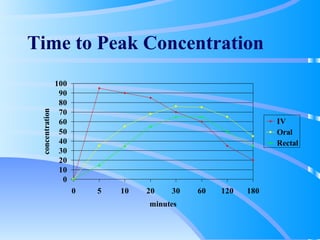










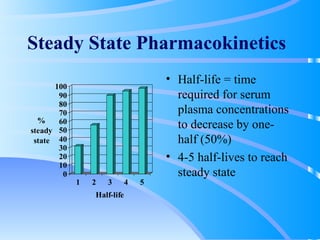
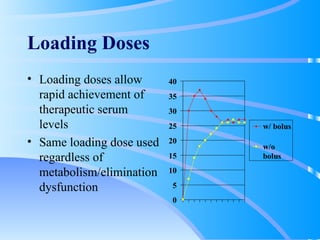






This document discusses clinical pharmacokinetics and pharmacodynamics. It defines pharmacokinetics as how the body affects a drug through absorption, distribution, metabolism and elimination. Factors like age can impact these processes in pediatric patients. It also discusses pharmacodynamics, how drugs act on the body, and how pharmacokinetics and pharmacodynamics together can help individualize drug therapy and decrease adverse effects.




























![Clinical Pharmacokinetics-II [dosing of drugs, tdm]](https://cdn.slidesharecdn.com/ss_thumbnails/clinicalpk-iidosingofdrugstdm-140217020047-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)
![Clinical Pharmacokinetics-I [half life, order of kinetics, steady state]](https://cdn.slidesharecdn.com/ss_thumbnails/clinicalpk-ihalflifeorderofkineticssteadystate-140217020044-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)

























































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)









