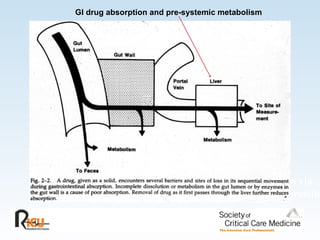
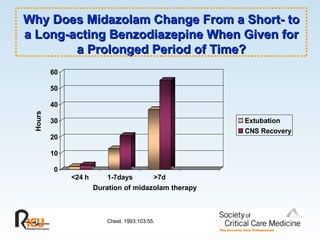
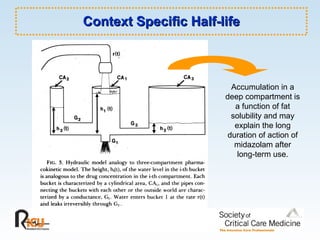
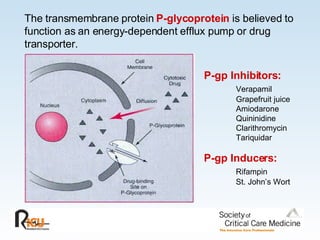
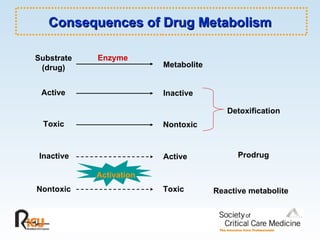
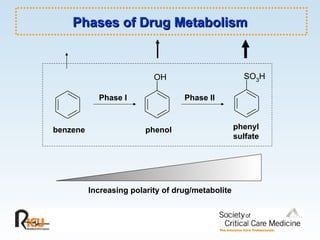
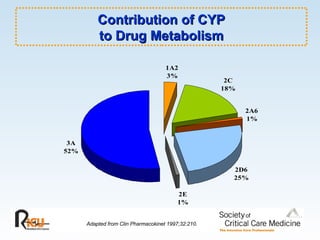
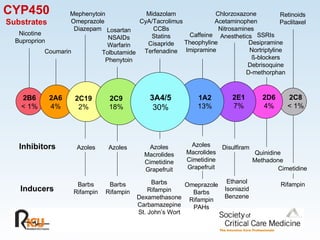
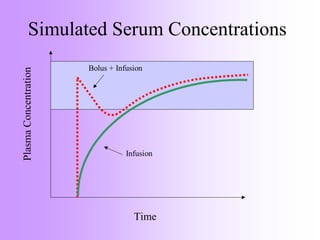
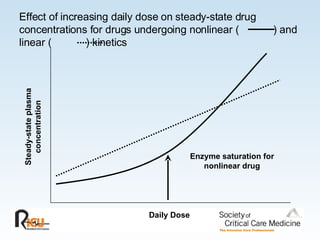
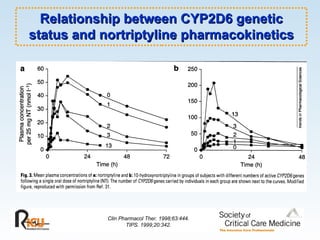
This document discusses principles of applied pharmacokinetics in critically ill adult patients. It covers topics like why pharmacokinetics is important, the four components of pharmacokinetics (absorption, distribution, metabolism, excretion), factors affecting drug absorption like routes of administration and the gastrointestinal tract, distribution and protein binding, phases of drug metabolism, and considerations for drug dosing in renal impairment. Case examples are provided to demonstrate practical applications of pharmacokinetic principles.










































































![Biotransformation_of_drug[ and contraindications of the 2].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/biotransformationofdrug2-240529164230-c1297680-thumbnail.jpg?width=640&height=640&fit=bounds)






















































