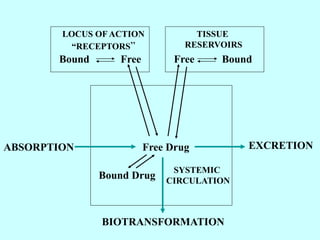
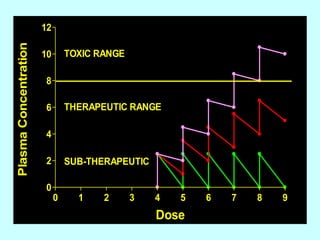
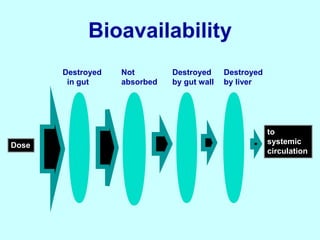
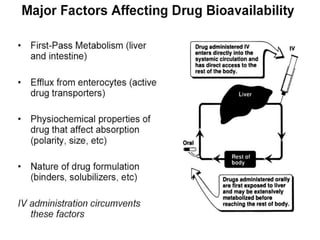
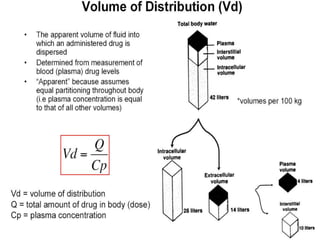
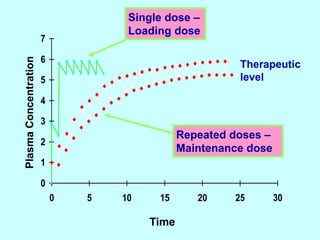
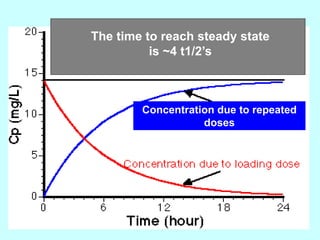
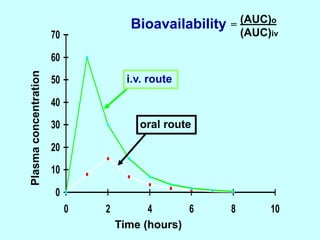
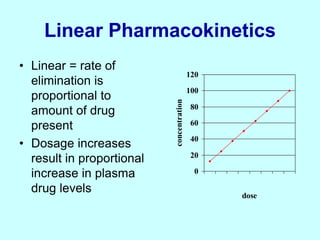
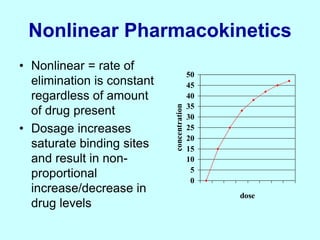
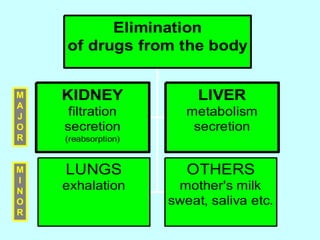
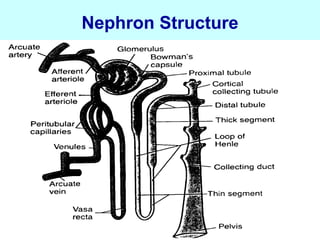
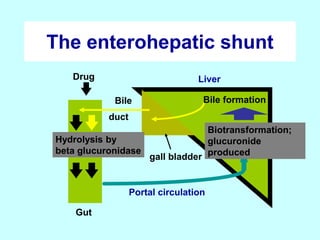
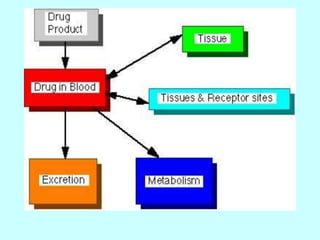
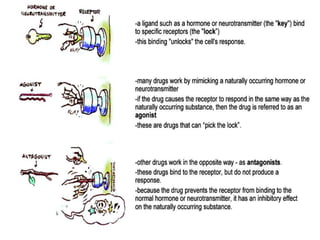
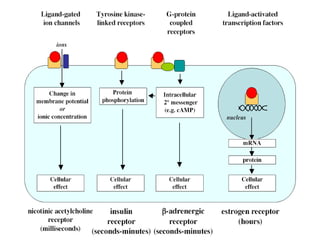
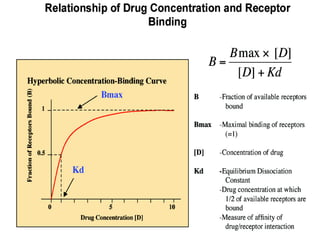
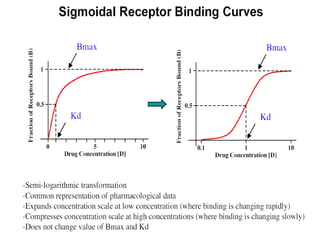
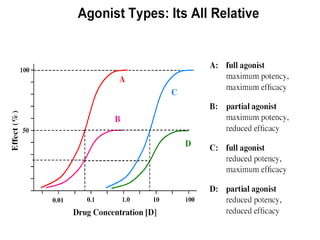
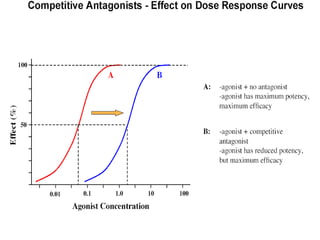
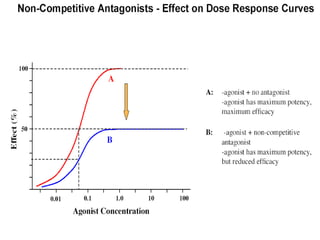
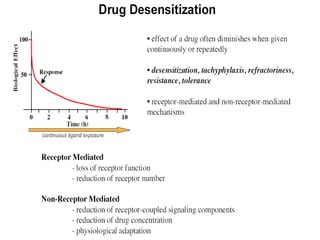
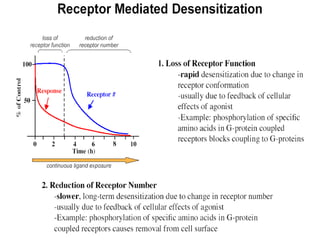
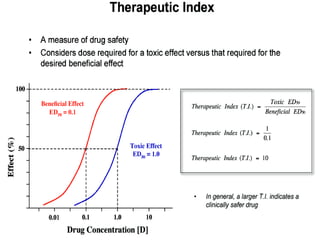
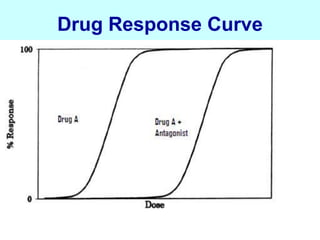
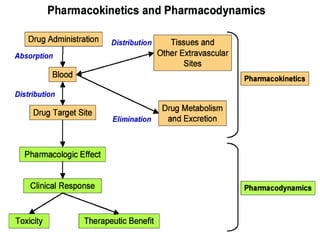
This document discusses pharmacokinetics (PK) and pharmacodynamics (PD). PK refers to what the body does to a drug, including absorption, distribution, metabolism and excretion. PD refers to what the drug does to the body, including its effects and mechanisms of action. The document covers topics such as drug receptors, agonists, antagonists, pharmacokinetic parameters like volume of distribution and half-life, and factors that affect a drug's behavior in the body like drug and patient characteristics.