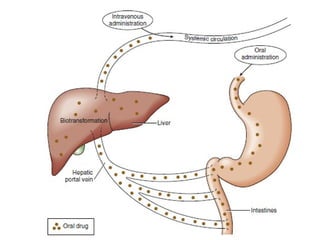
Pharmacology describes a drug's journey through the body and its effects. Pharmacokinetics involves absorption, distribution, metabolism and excretion of a drug. Absorption depends on route of entry and bioavailability. Distribution is based on volume of distribution. Metabolism occurs mainly in the liver through phase I and II reactions. Excretion eliminates drugs through kidneys or bile. Pharmacodynamics studies how drugs cause their effects by interacting with receptors.














































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)



