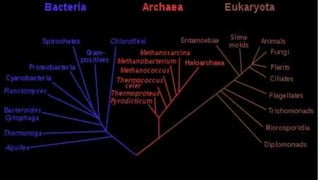
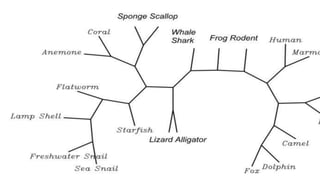
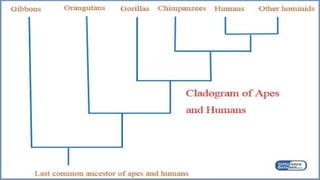
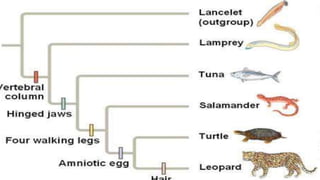
A phylogenetic tree is a branching diagram representing the evolutionary relationships among biological species based on genetic or physical characteristics. It can be categorized into rooted and unrooted trees, with further distinctions made between bifurcating and multifurcating trees, as well as labeled and unlabeled trees. Although providing insights into evolutionary history, phylogenetic trees have limitations, including the potential for inaccuracies due to factors like genetic recombination and the noise in data, emphasizing the need for multi-gene analysis and caution in interpretation.











![Spindle diagram
A spindle diagram, or bubble diagram, is often called a
romerogram, after its popularisation by the American
palaeontologist Alfred Romer.
It represents taxonomic diversity (horizontal width)
against geological time (vertical axis) in order to reflect
the variation of abundance of various taxa through time.
However, a spindle diagram is not an evolutionary
tree the taxonomic spindles obscure the actual
relationships of the parent taxon to the daughter
taxon[16] and have the disadvantage of involving
the paraphyly of the parental group. This type of diagram
is no longer used in the form originally proposed.](https://image.slidesharecdn.com/phylogenetictree-210611075412/85/Phylogenetic-tree-and-it-s-types-15-320.jpg)





















































































