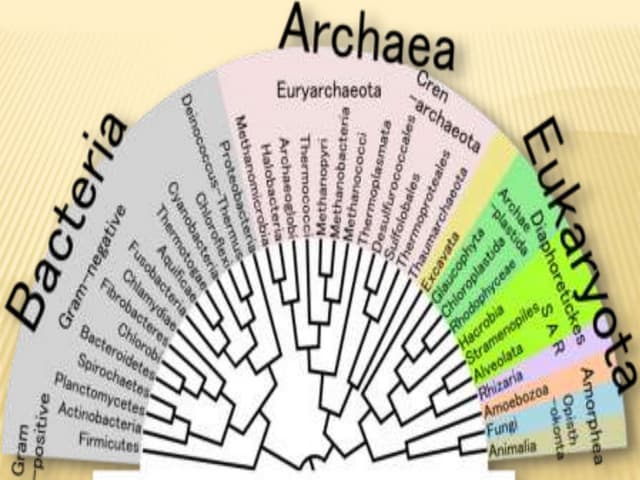
This document provides an introduction to phylogenetic analysis. It discusses key terminology like taxonomy, phylogeny, and phylogenetics. The document explains that phylogenetic analysis is used to study evolutionary relationships between organisms and sequences that are derived from a common ancestor. It also summarizes several commonly used methods for constructing phylogenetic trees from sequence data, including distance matrix methods, parsimony methods, and maximum likelihood. The document outlines factors to consider when selecting sequences for phylogenetic analysis and some known problems with multiple sequence alignments.