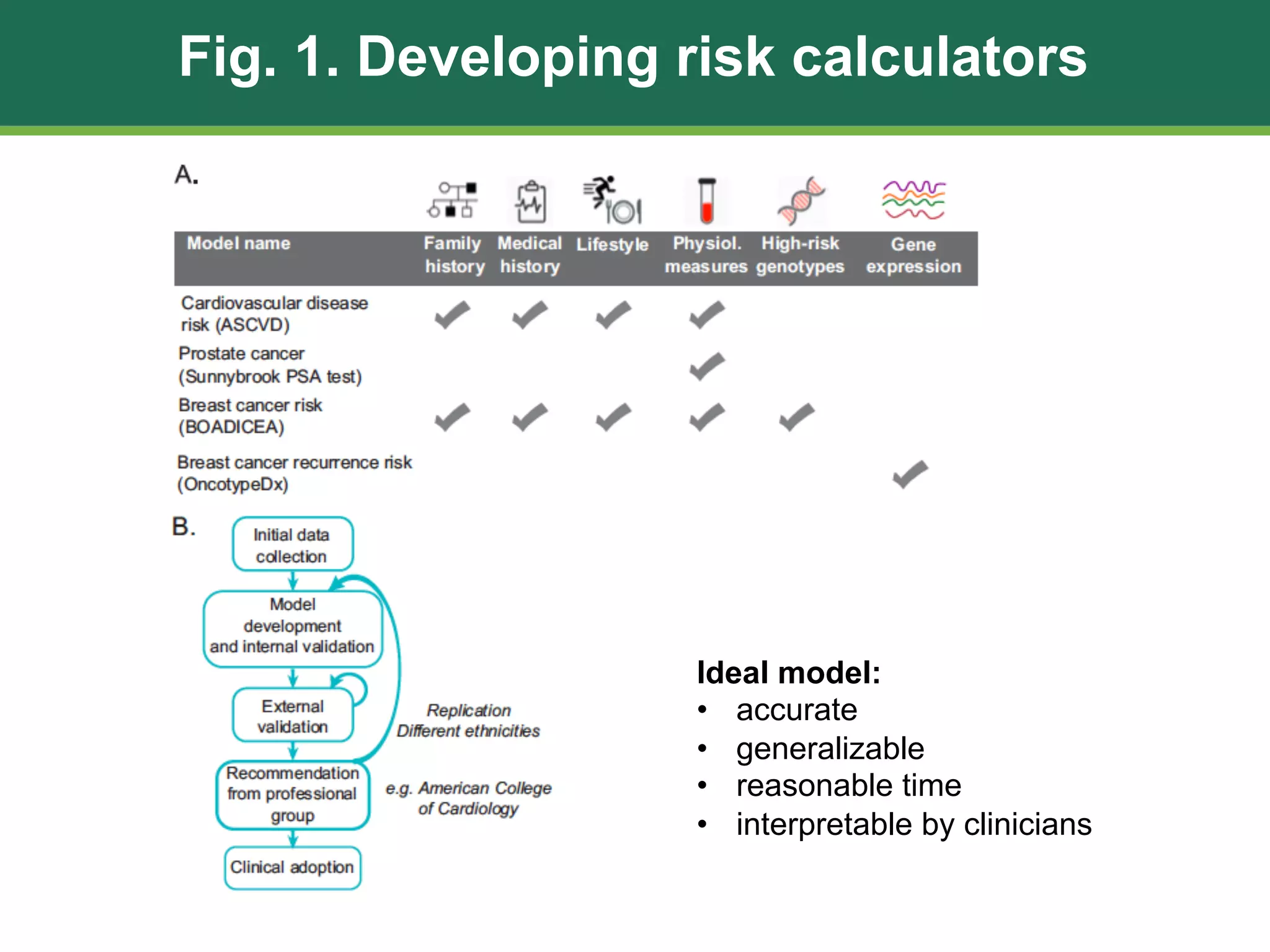
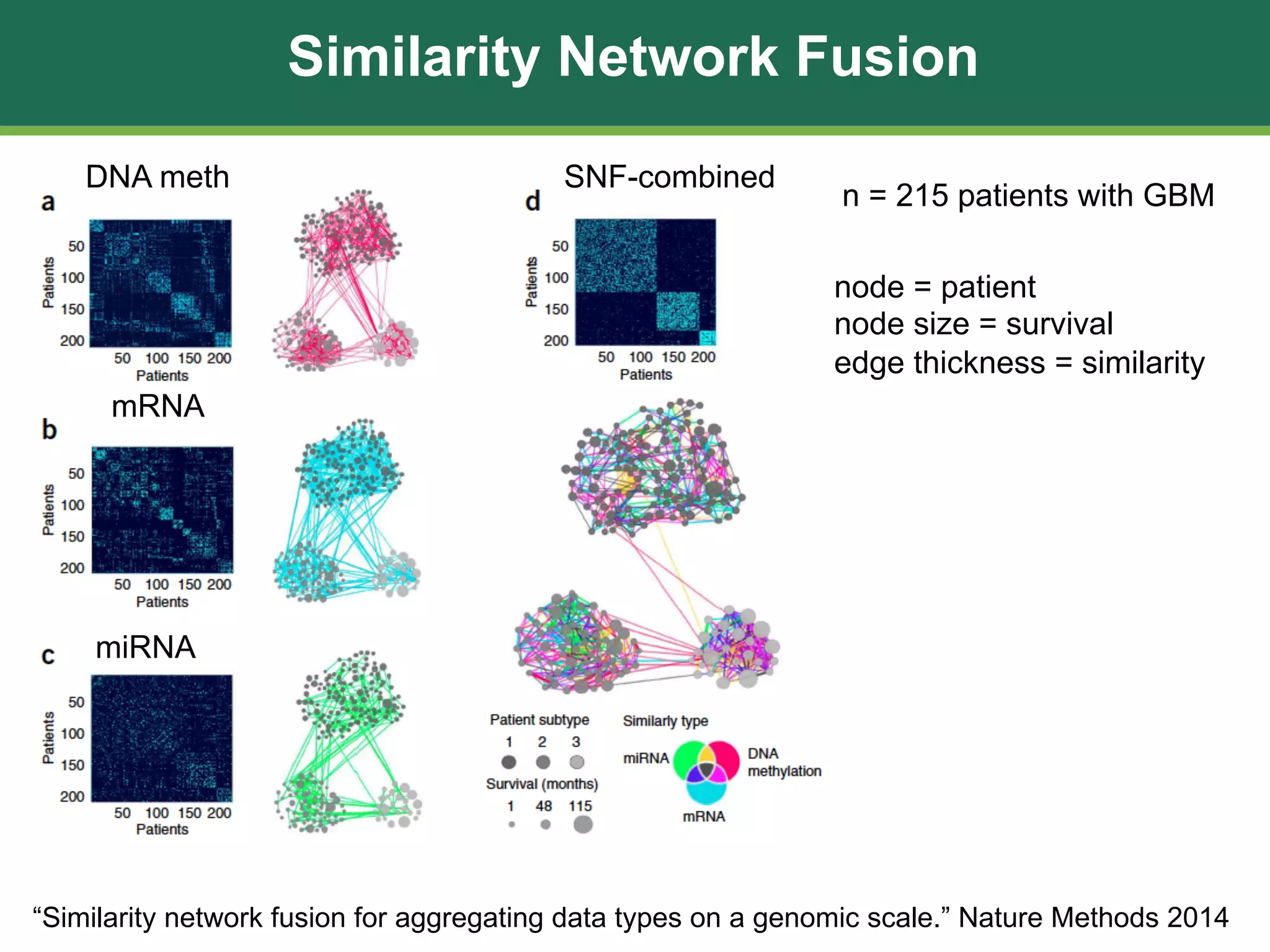
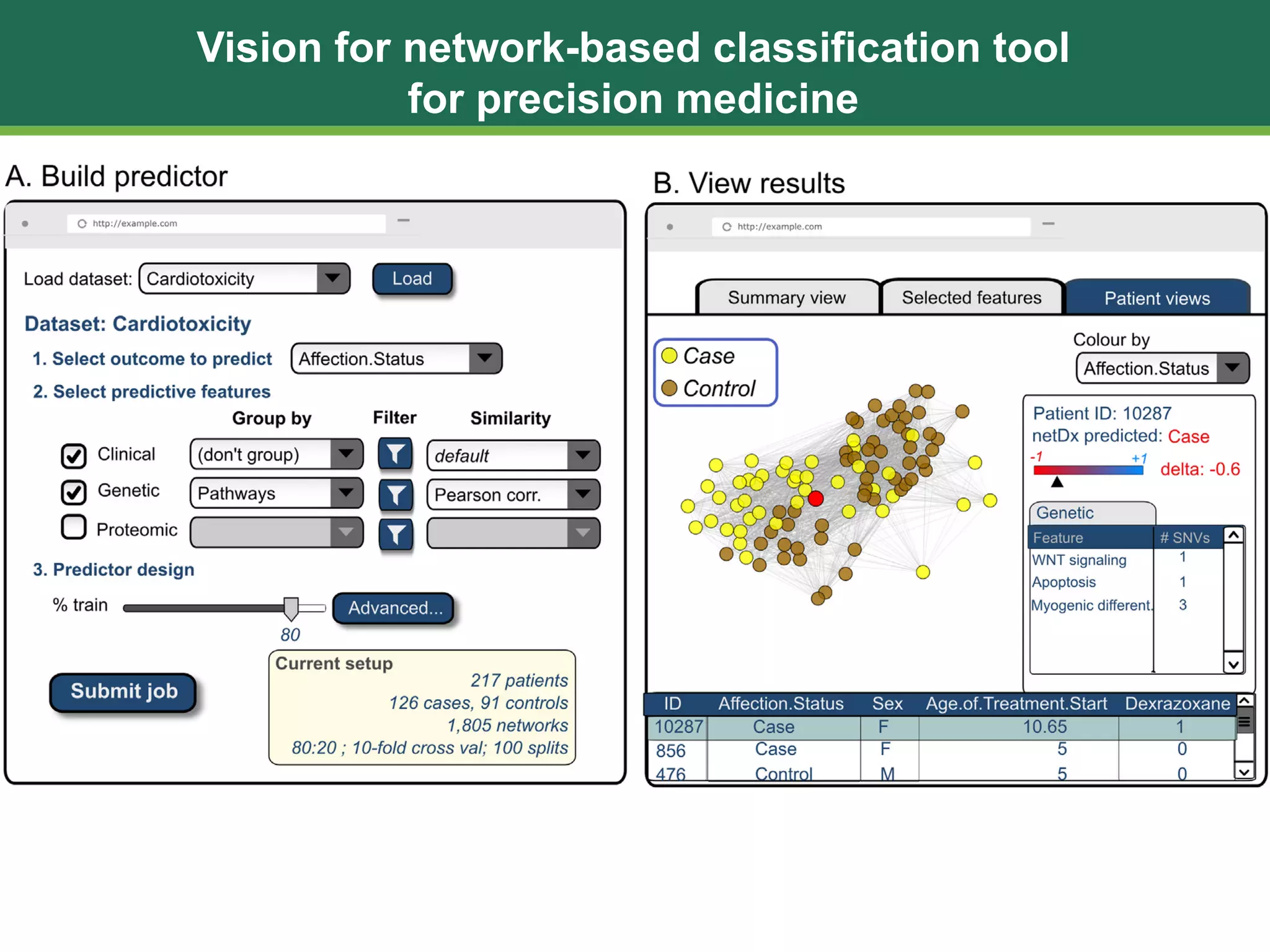
The document discusses patient similarity networks (PSNs) as an emerging method in precision medicine for building predictive risk models by integrating diverse data types while maintaining patient privacy. It highlights two frameworks, similarity network fusion and netdx, which successfully implement PSN, and explores the advantages and challenges associated with this approach. The author proposes that PSNs can improve clinical decision-making through better visualization and intuitive patient grouping, despite facing obstacles like large data sizes and the need for effective feature selection.