









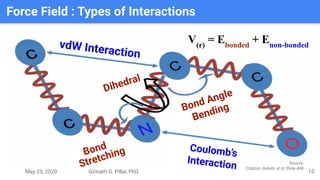
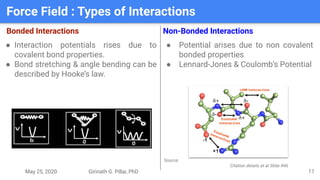

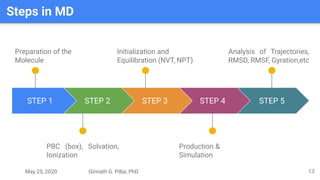
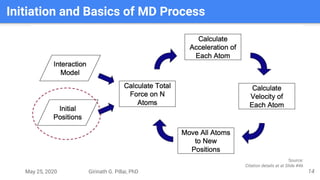








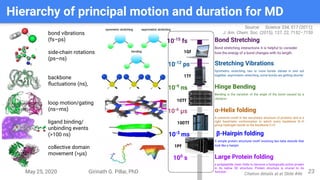




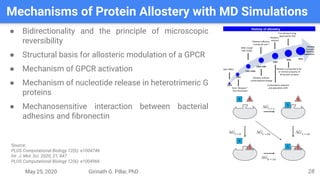


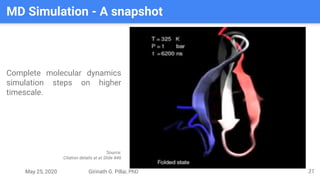

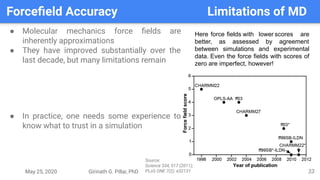





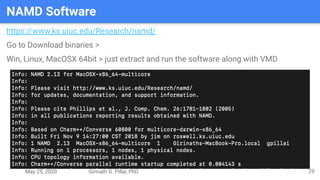


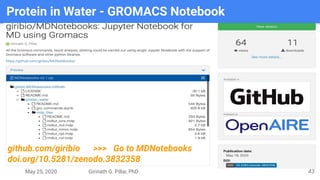
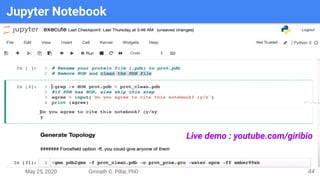


The presentation by Girinath G. Pillai provides a comprehensive overview of molecular dynamics (MD), covering its fundamentals, implementation steps, and applications in analyzing atomic and molecular behavior. Key topics discussed include the mechanics of MD simulations, types of force fields, the importance of periodic boundary conditions, and strategies for calculating energetics while managing computational demands. The document emphasizes the significance of MD in fields such as protein folding, ligand interactions, and estimating thermodynamic properties.



















































































