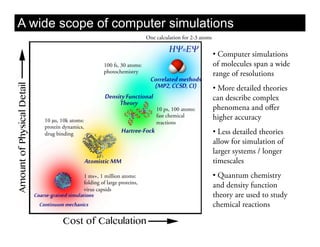
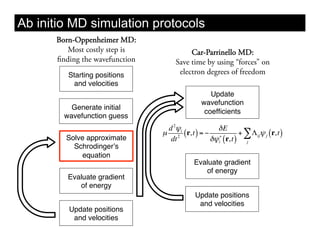
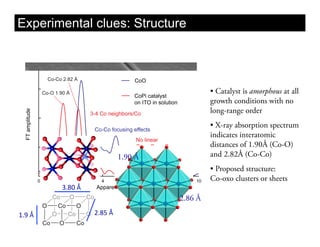
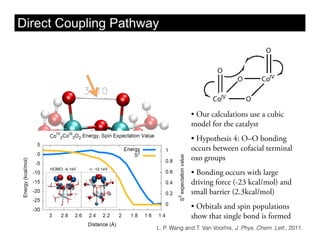
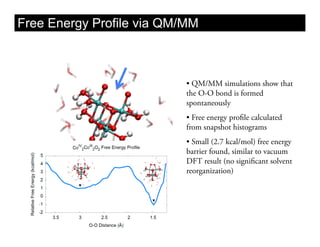
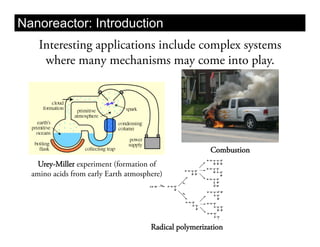
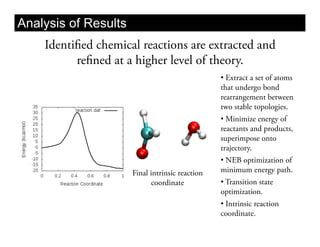
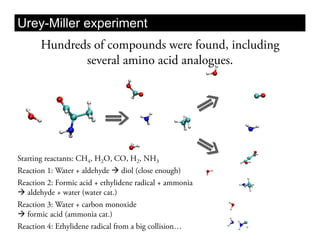
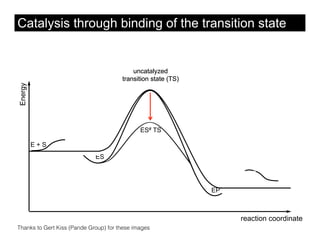
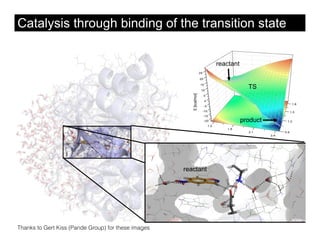
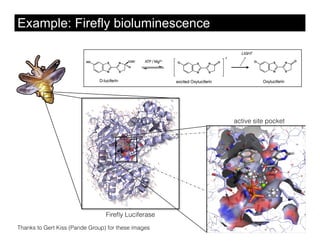
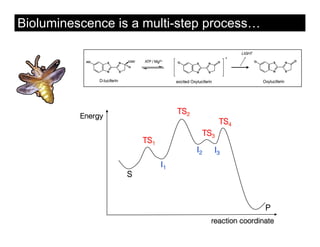
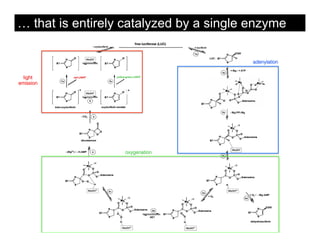
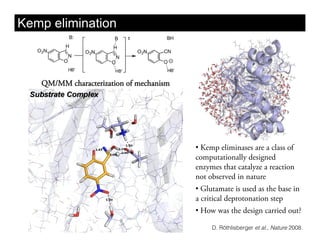
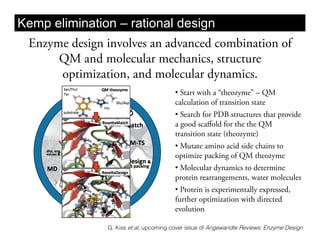
This document discusses ab initio molecular dynamics simulation methods. It provides an overview of different simulation techniques that range from fully quantum to mixed quantum-classical approaches. These methods allow researchers to study molecular phenomena with varying degrees of accuracy and system sizes. The document also outlines key concepts like the Schrodinger equation and Born-Oppenheimer approximation that are fundamental to these simulation approaches.