Downloaded 296 times



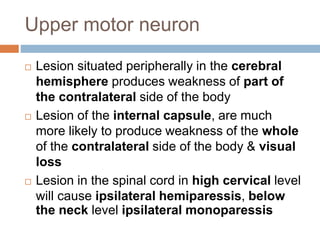















This document summarizes several motor neuron diseases: - Upper motor neuron lesions are characterized by increased tone, weakness in anti-gravity muscles, and increased reflexes while lower motor neuron lesions show wasting, fasciculation, decreased tone and reflexes. - Amyotrophic lateral sclerosis (ALS) involves both upper and lower motor neurons leading to weakness and atrophy. Median survival is 3 years. - Spinal muscular atrophy is an autosomal recessive disorder caused by a defect on chromosome 5q that leads to loss of anterior horn cells and progressive limb and respiratory weakness.























































































