Downloaded 68 times
















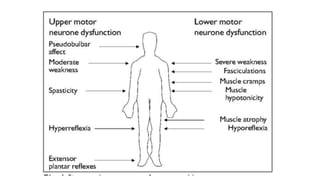



























This document provides information on motor neuron disease and amyotrophic lateral sclerosis (ALS) in particular. It discusses the pathology, classification, clinical manifestations, investigations, differential diagnosis and treatment of motor neuron diseases with a focus on ALS. ALS is characterized by the degeneration of both upper and lower motor neurons leading to muscle weakness, atrophy and fasciculations. Riluzole and edaravone are approved treatments that can modestly prolong survival.




























































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)













