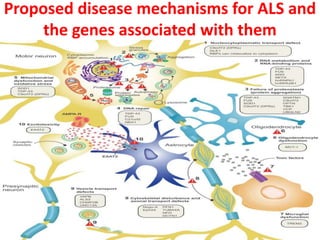
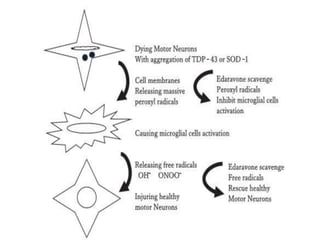
1) Recent advances in understanding the pathophysiology of motor neuron disease include insights into excitotoxicity, oxidative stress, mitochondrial defects, impaired axonal transport, protein aggregation, inflammation, and neurotrophic factor deficits.
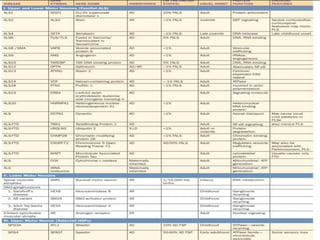
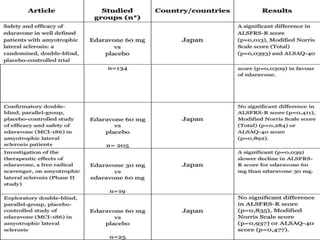
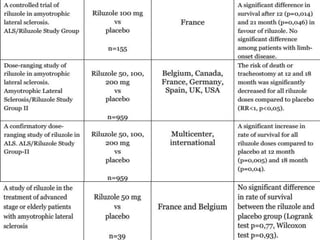
2) Riluzole remains the only FDA-approved drug shown to modestly prolong survival for patients with ALS, though Edaravone may also provide benefits for certain subgroups. Experimental therapies targeting genes, antioxidants, neurotrophic factors, and other mechanisms are under investigation.
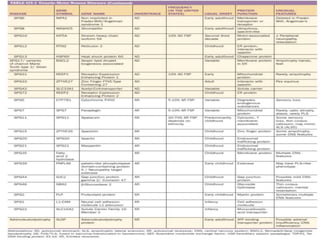
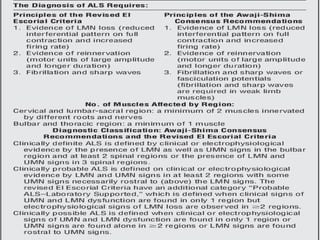
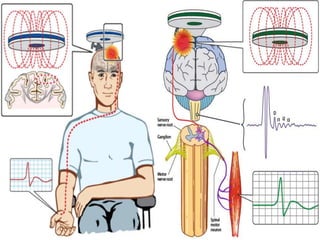
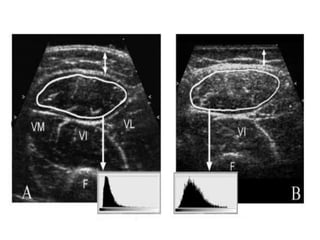
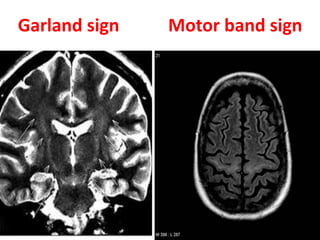
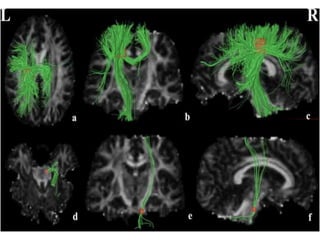
3) Making an accurate diagnosis involves evaluating the patient's history, physical exam, electrodiagnostic testing, imaging, and sometimes genetic or biomarker analysis to differentiate ALS from other conditions.






































































































![DUAL AND TRIPLE ANTITHROMBOTIC THERAPY FOR SECONDARY STROKE [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/dualandtripleantithrombotictherapyforsecondarystrokeautosaved-230904113552-c3502b37-thumbnail.jpg?width=640&height=640&fit=bounds)

































