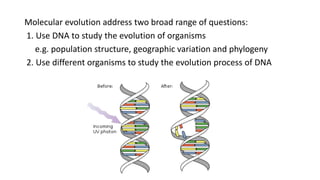
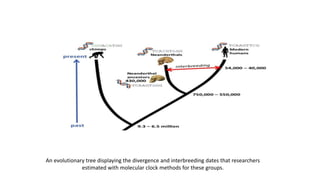
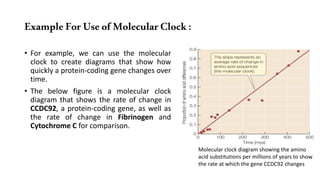
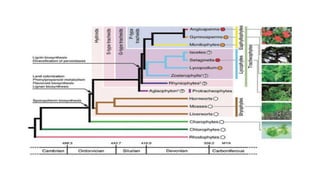
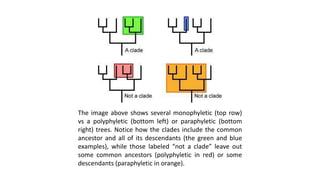
The document discusses the applications of bioinformatics in evolutionary studies, detailing concepts such as molecular phylogeny, the molecular clock hypothesis, and Kimura's theory. It highlights the role of bioinformatics tools in analyzing genetic sequences to reconstruct evolutionary relationships, understand molecular evolution, and visualize phylogenetic trees. Additionally, it elaborates on various types of phylogenetic trees and their uses in studying species diversity, conservation, and pathogen origins.