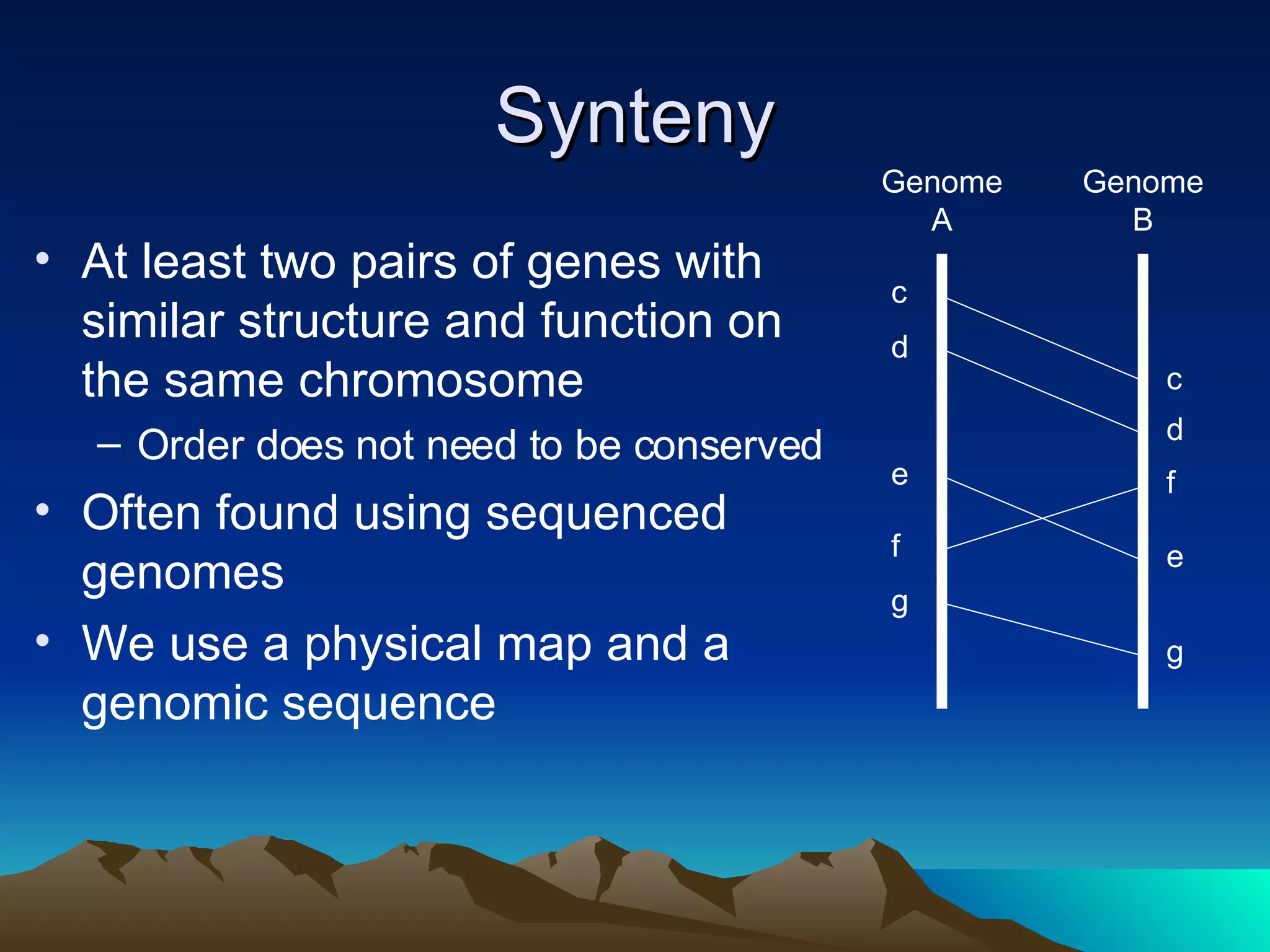
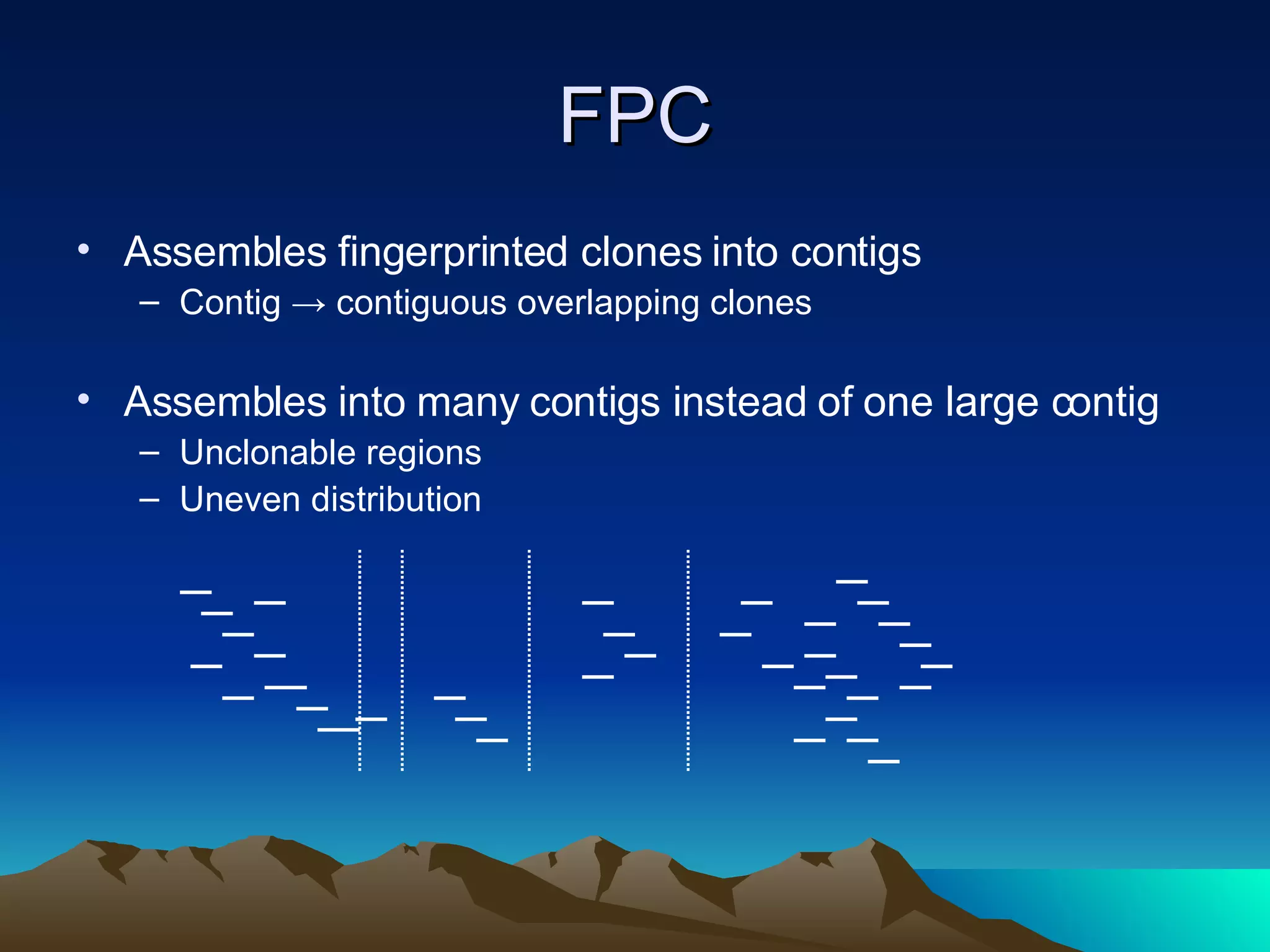
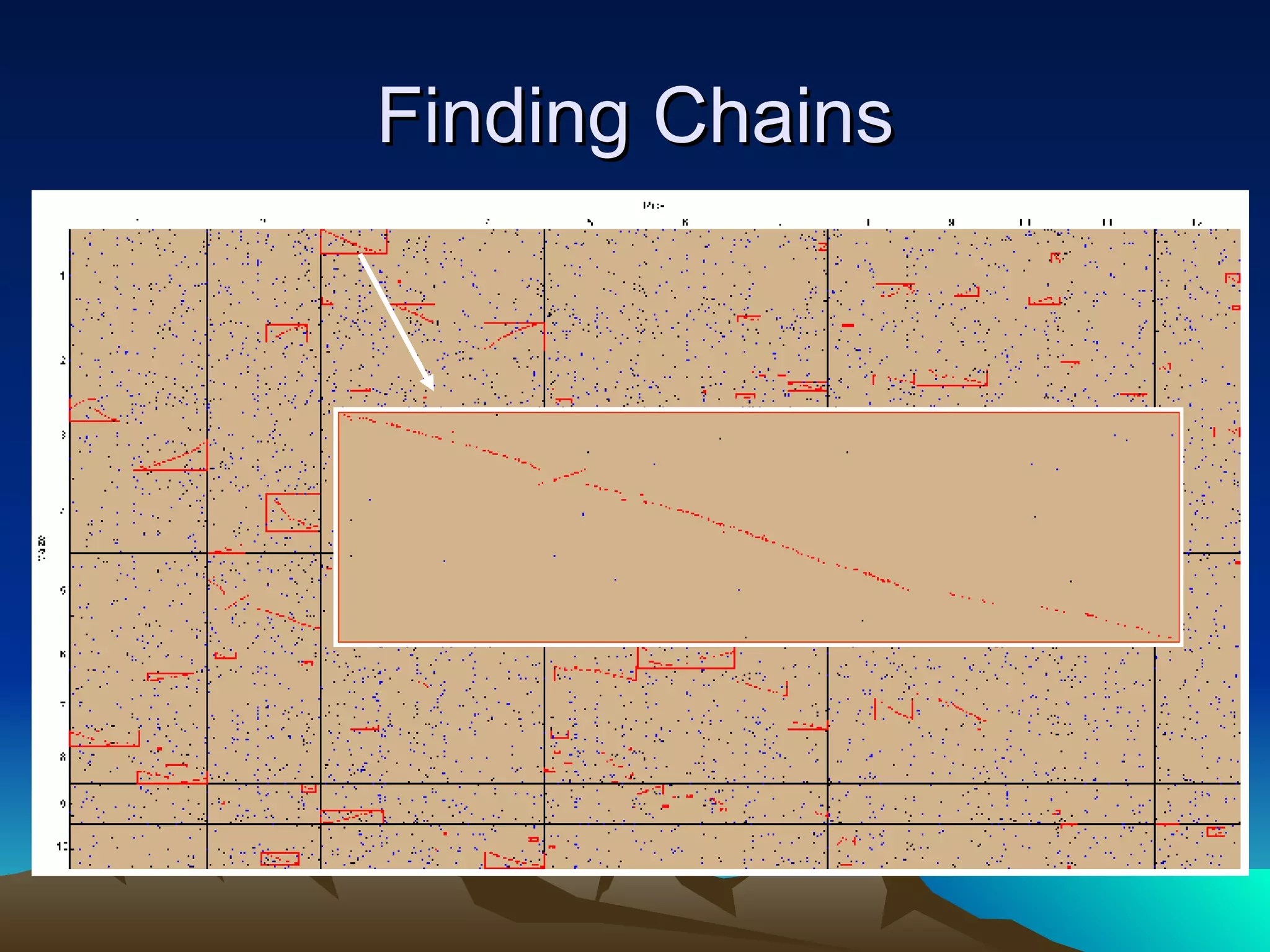
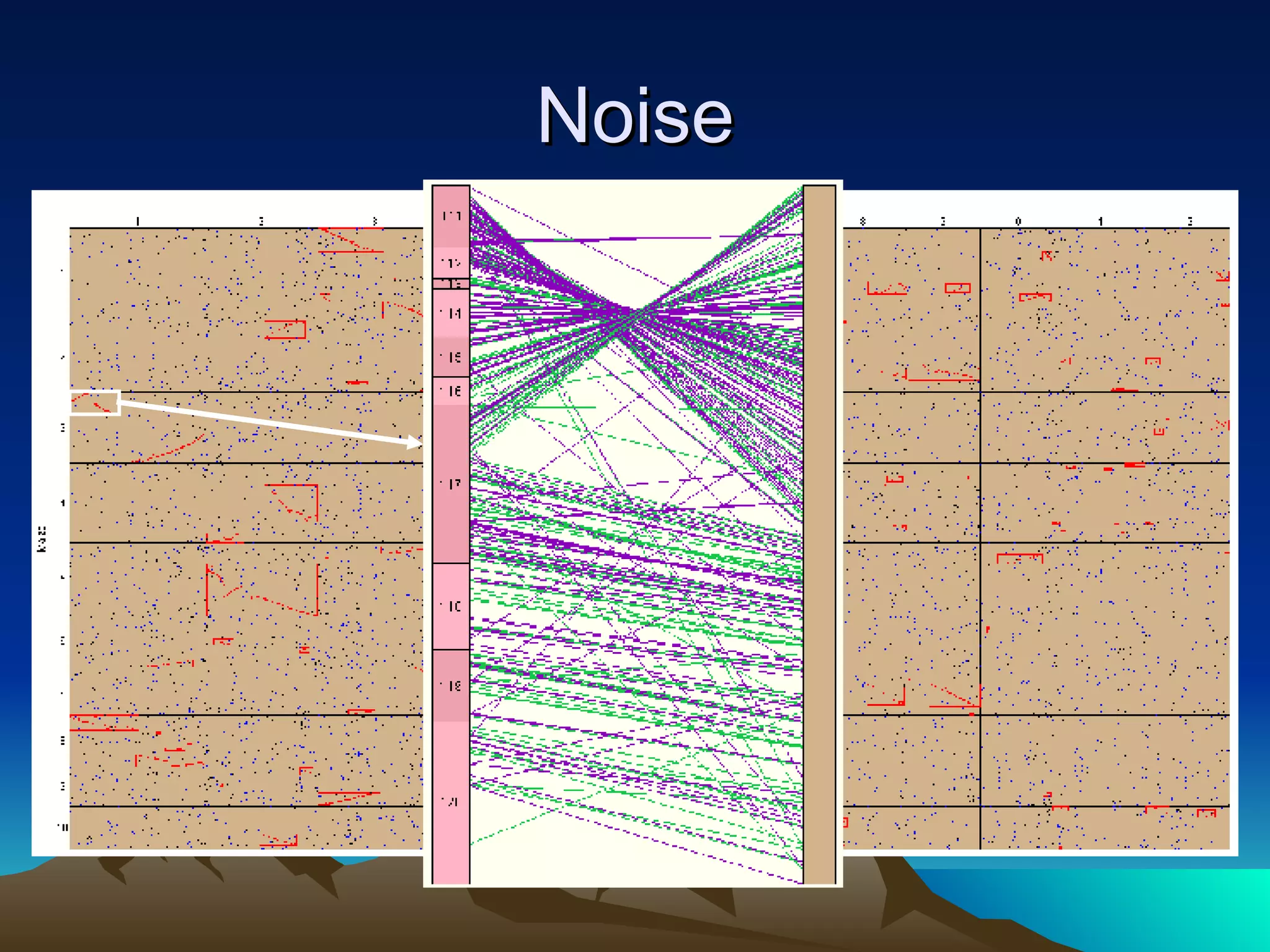
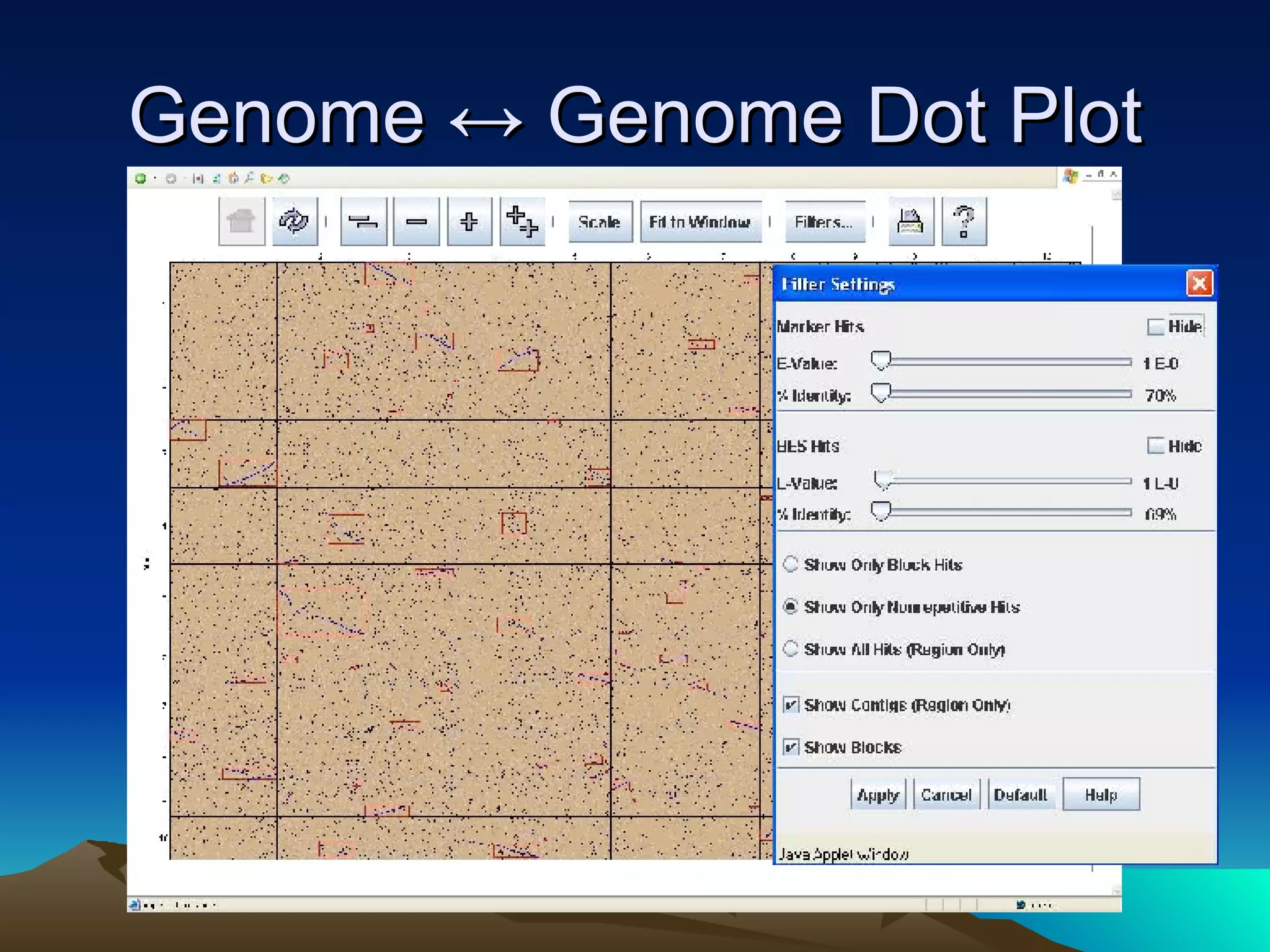
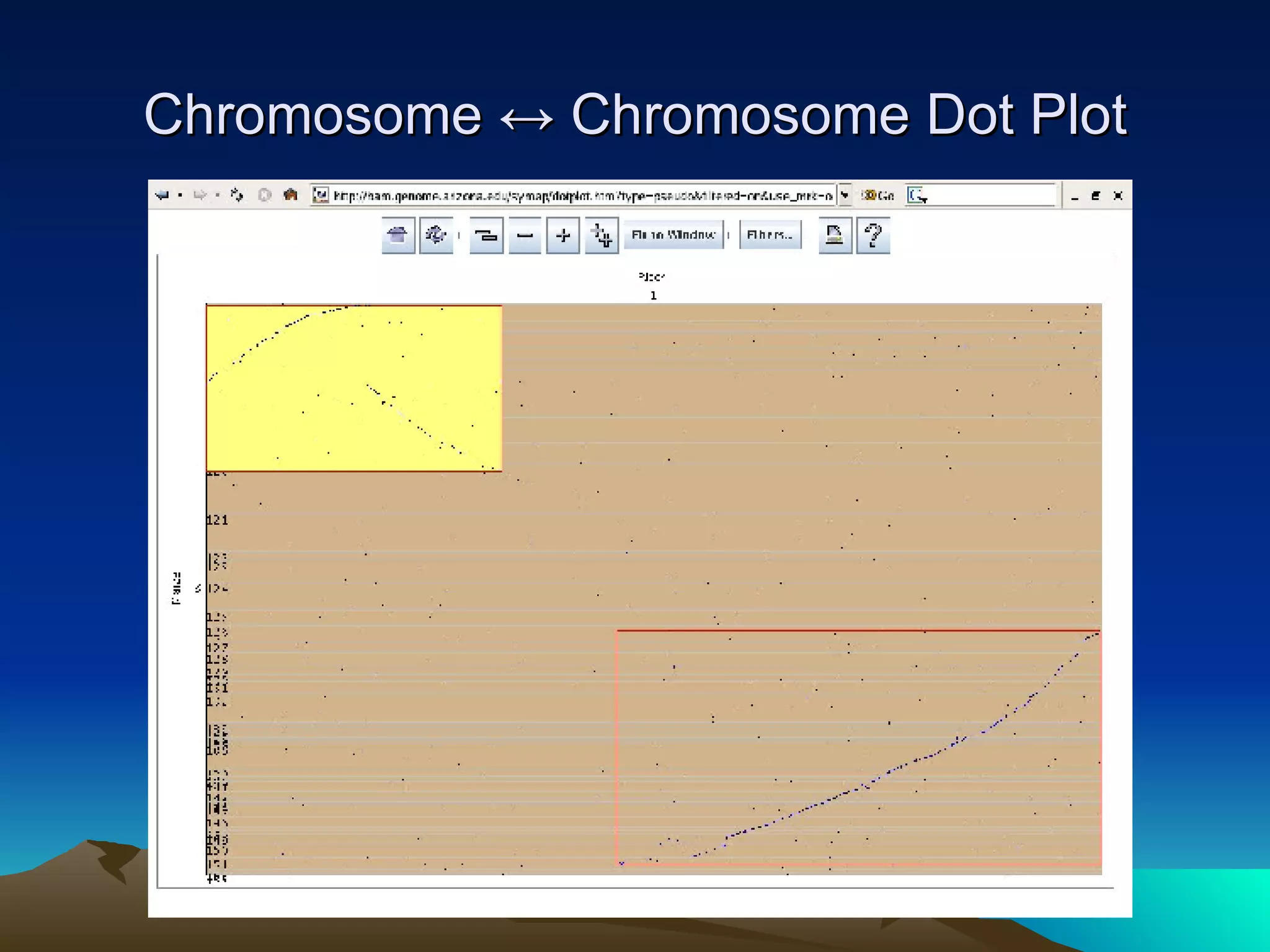
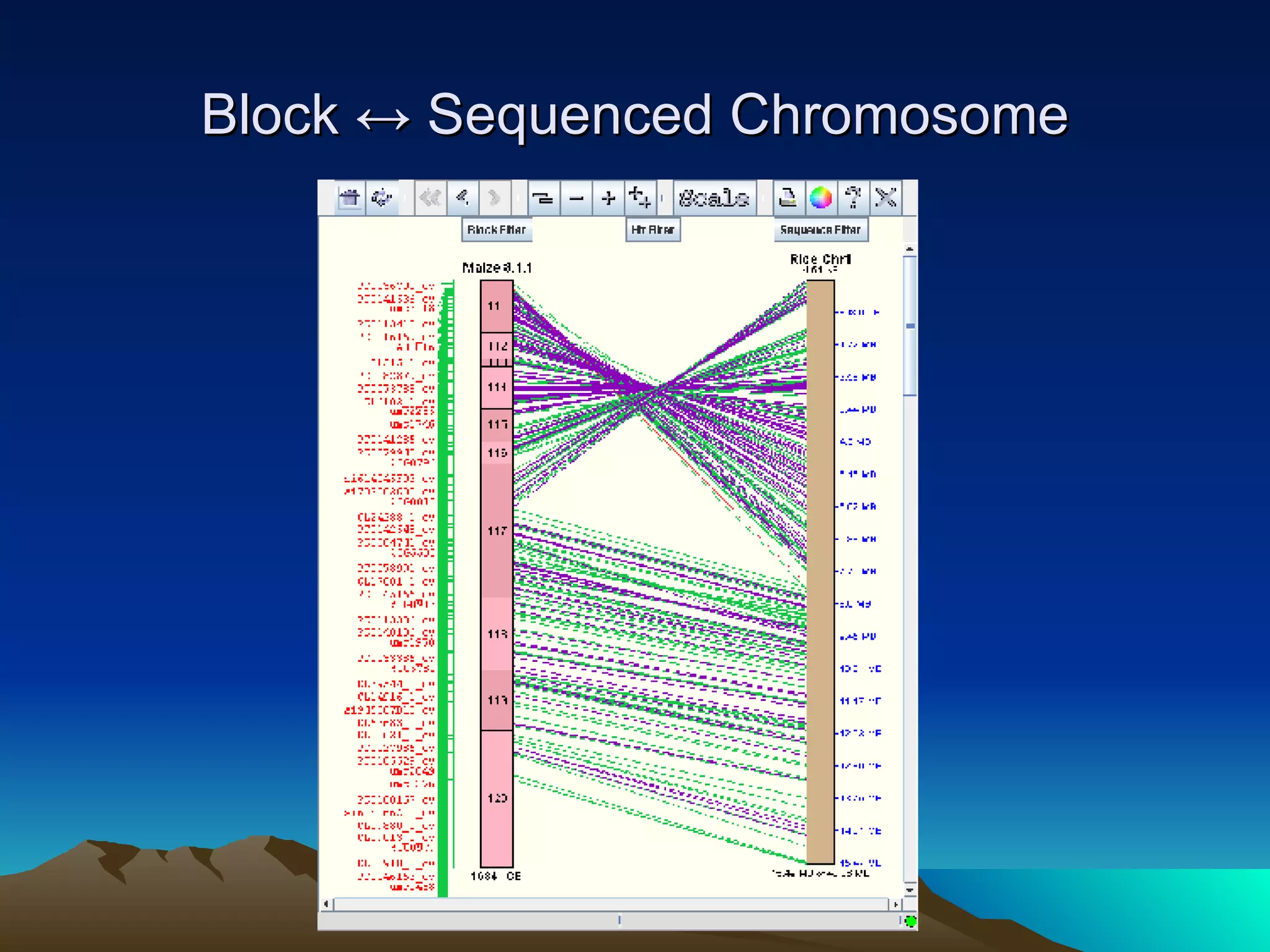
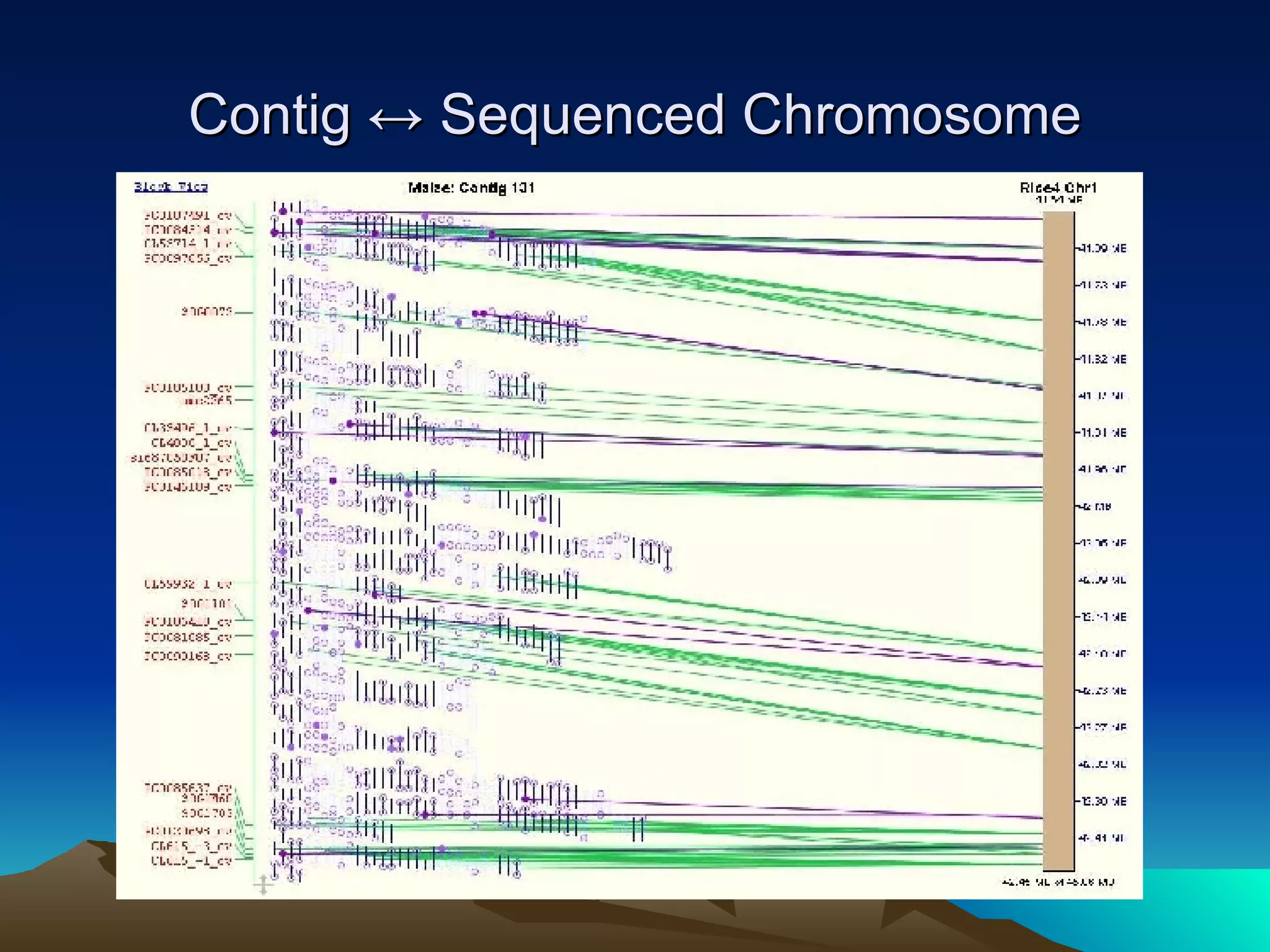
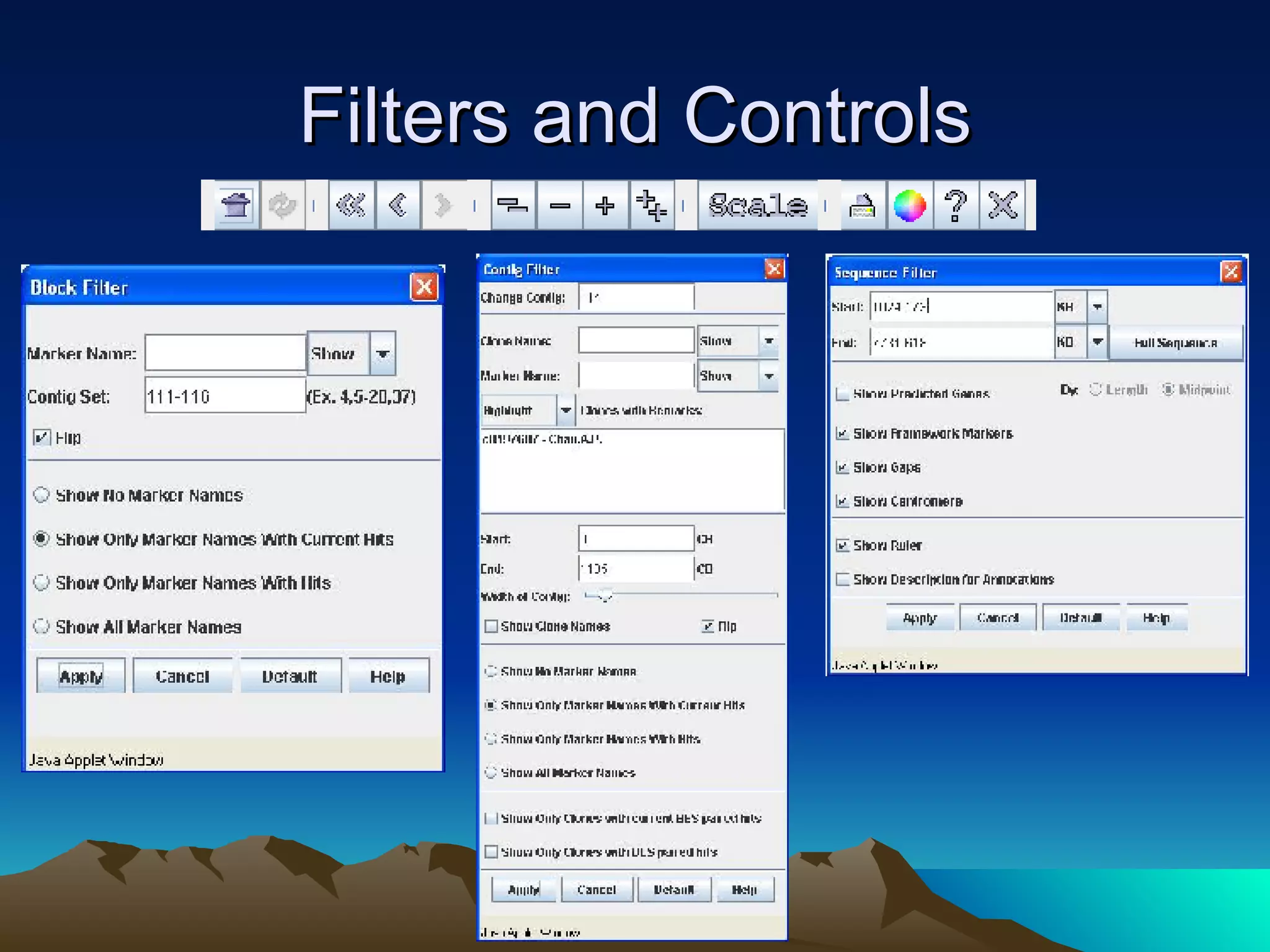
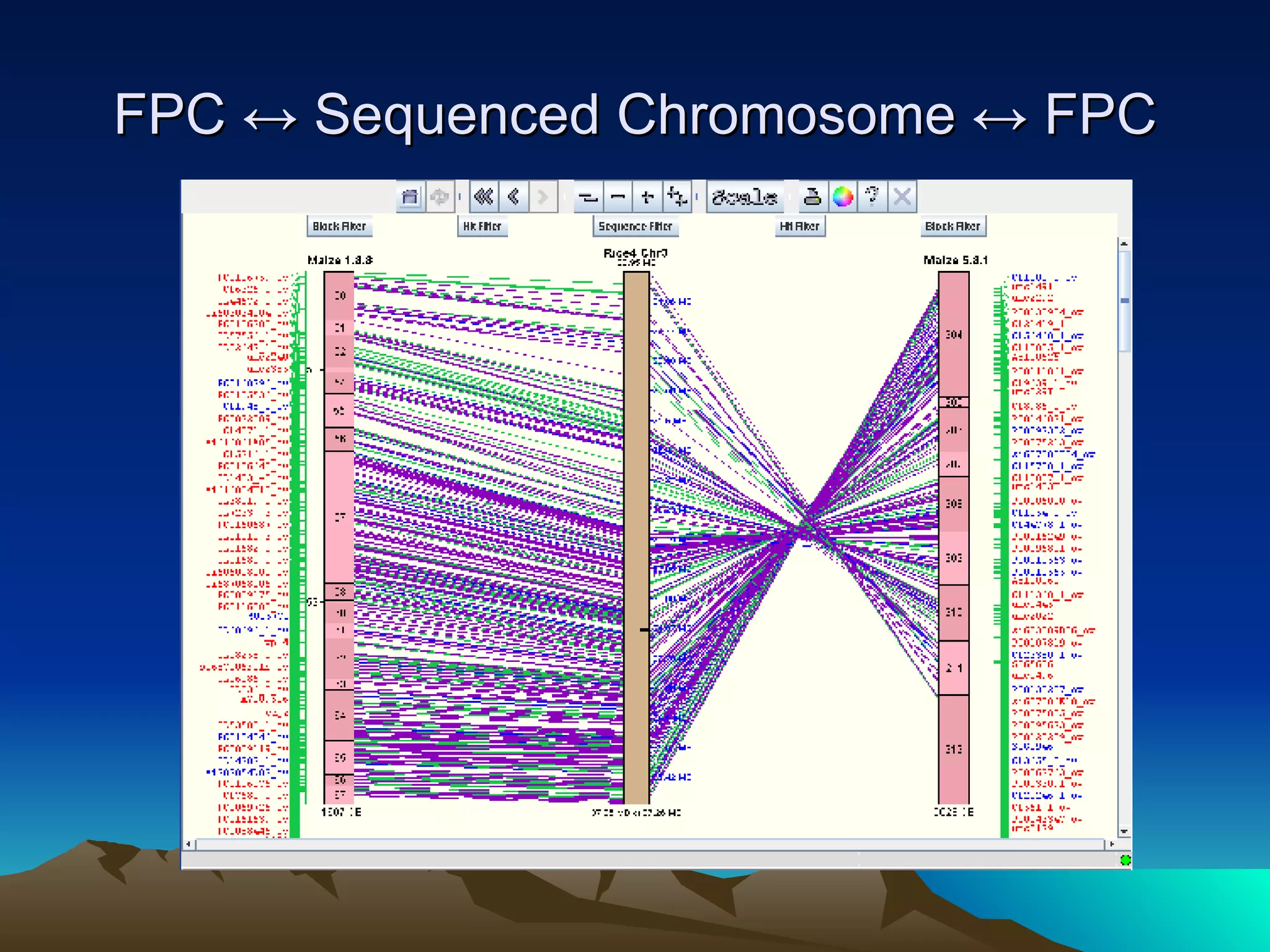
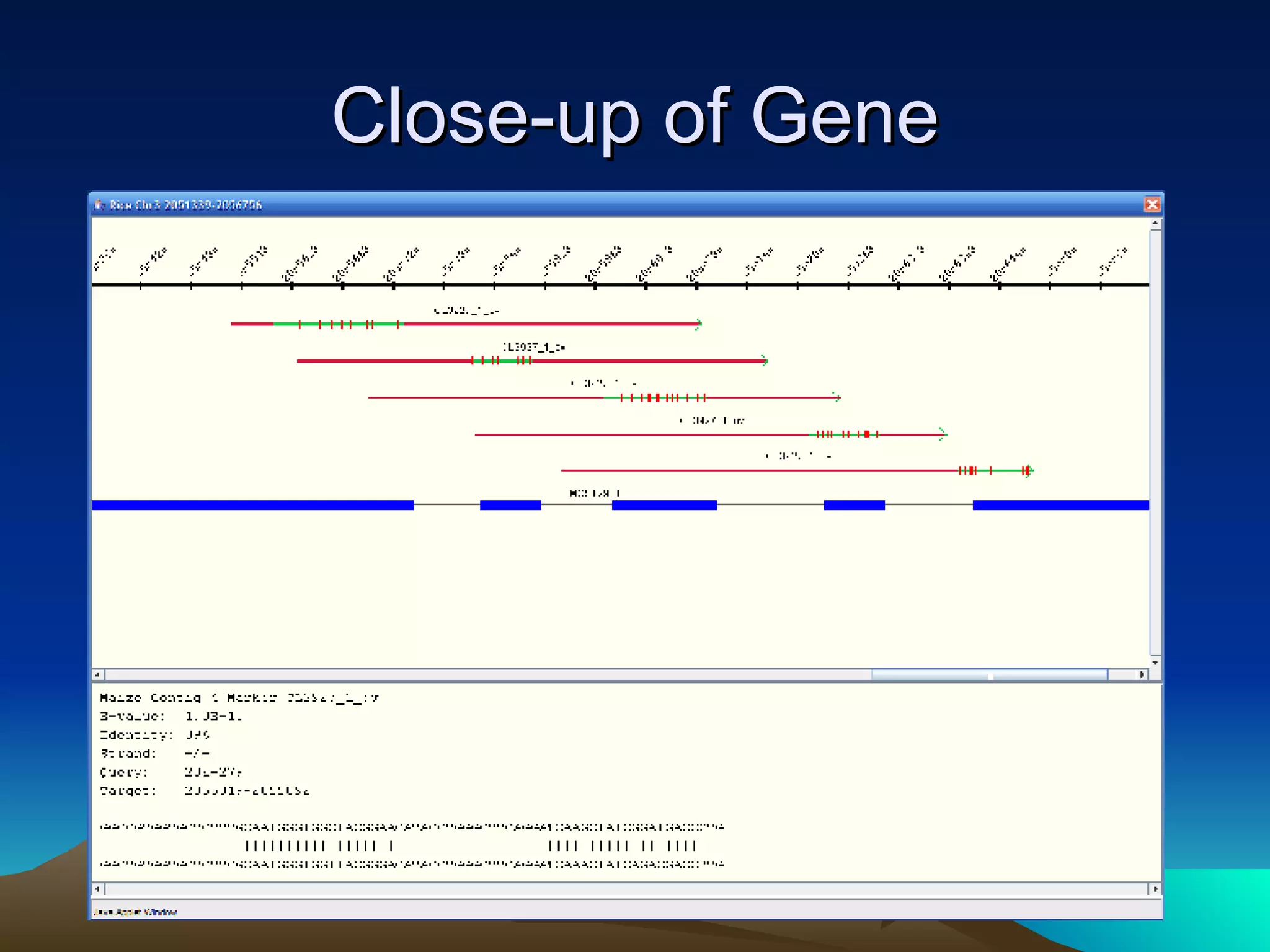
SyMAP is a synteny mapping and analysis program that compares genomes of different species to understand gene function and evolutionary history. It finds synteny blocks between a physical map (e.g. FPC map) and a sequenced genome using anchors like markers and BAC end sequences. The algorithm uses a directed acyclic graph and dynamic programming to order anchors into synteny chains while allowing for errors and rearrangements. SyMAP displays synteny results through interactive views and aids in tasks like correcting BAC clone end assignments. It has been applied to several plant genome projects.






































































![Coded Agents – with UiPath SDK + LangGraph [Virtual Hands-on Workshop]](https://cdn.slidesharecdn.com/ss_thumbnails/codedagentsdeck-251215155422-5497c599-thumbnail.jpg?width=640&height=640&fit=bounds)










