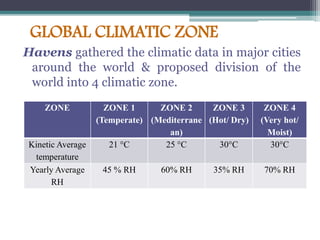
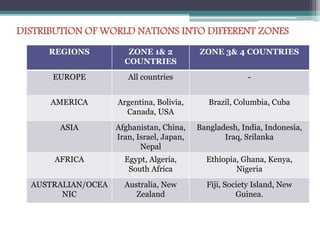
The document elaborates on the International Conference on Harmonization (ICH), which aims to harmonize technical requirements for pharmaceutical registration globally to enhance the safety, efficacy, and quality of medicines. It discusses the structure of ICH, including key regulatory bodies from Europe, Japan, and the USA, as well as the categories of guidelines that have been developed, such as quality, safety, and efficacy topics. Additionally, it outlines the importance of stability testing for drug substances and products, including revised storage conditions and commitment to ongoing stability studies.