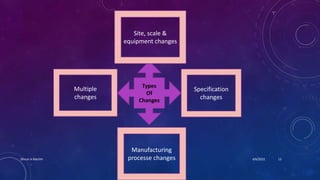
The document is a presentation on post-approval changes to bulk active chemicals. It discusses FDA guidance called BACPAC (Bulk Active Chemicals & Post Approval Changes) which provides recommendations for post-approval changes to drug substance synthesis, including site, scale and equipment changes as well as specification and manufacturing process changes. The guidance covers assessing equivalence after changes and determining the appropriate reporting category based on the potential effects of changes.