Downloaded 5,543 times
































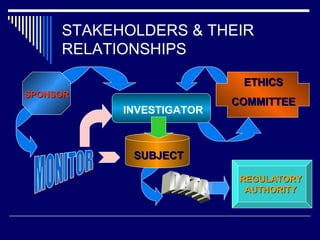















The document provides an overview of ICH GCP (International Council for Harmonisation Good Clinical Practice) guidelines. ICH GCP guidelines were developed to harmonize clinical trial standards and processes across regions. They establish international ethical and scientific quality standards for designing, conducting, and reporting clinical research involving human subjects. Adherence to ICH GCP provides public assurance that the rights, safety, and well-being of clinical trial subjects are protected.















































