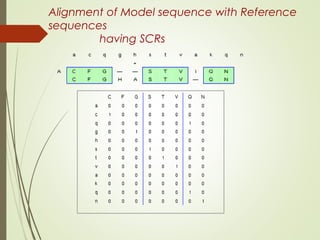
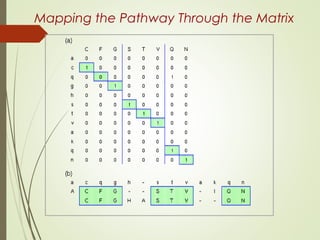
Homology modeling is a technique used to generate a 3D model of a protein's structure using an evolutionary related template protein with a known structure. The main steps are template recognition, sequence alignment, determining conserved regions, backbone generation, building loops, side chain refinement, and structure validation. The accuracy of the model depends on selecting a suitable template and proper alignment between the target and template sequences.