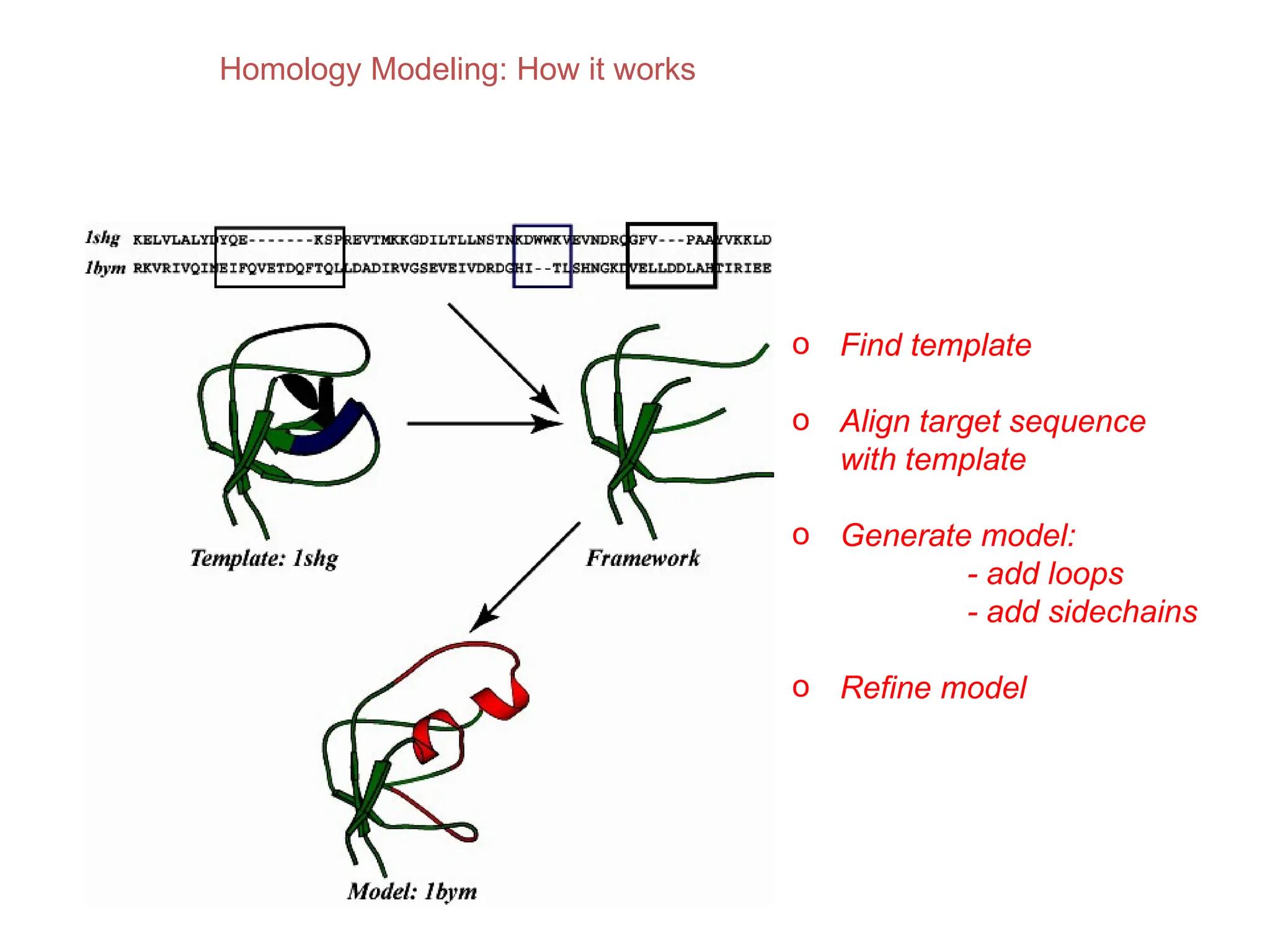
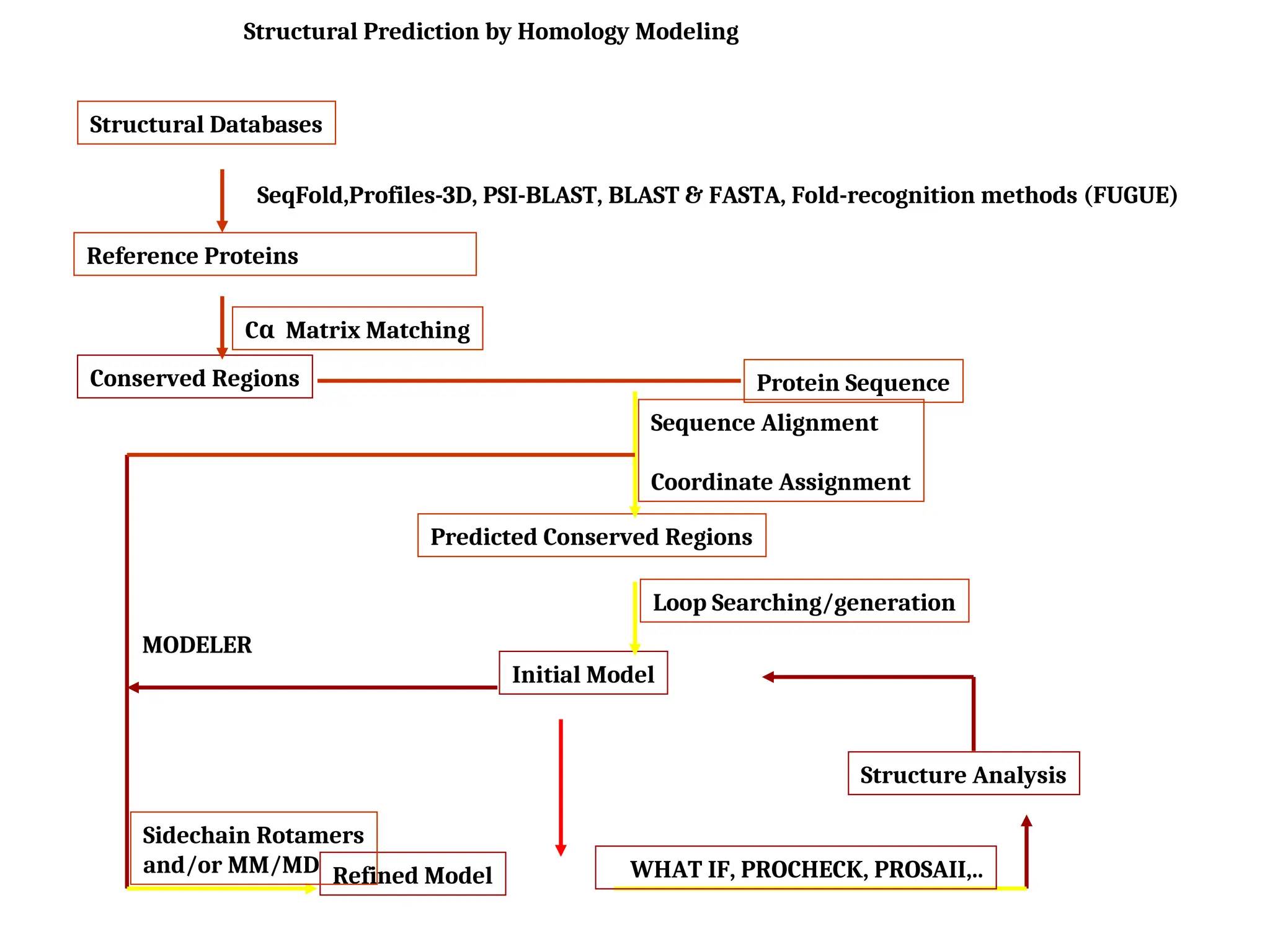
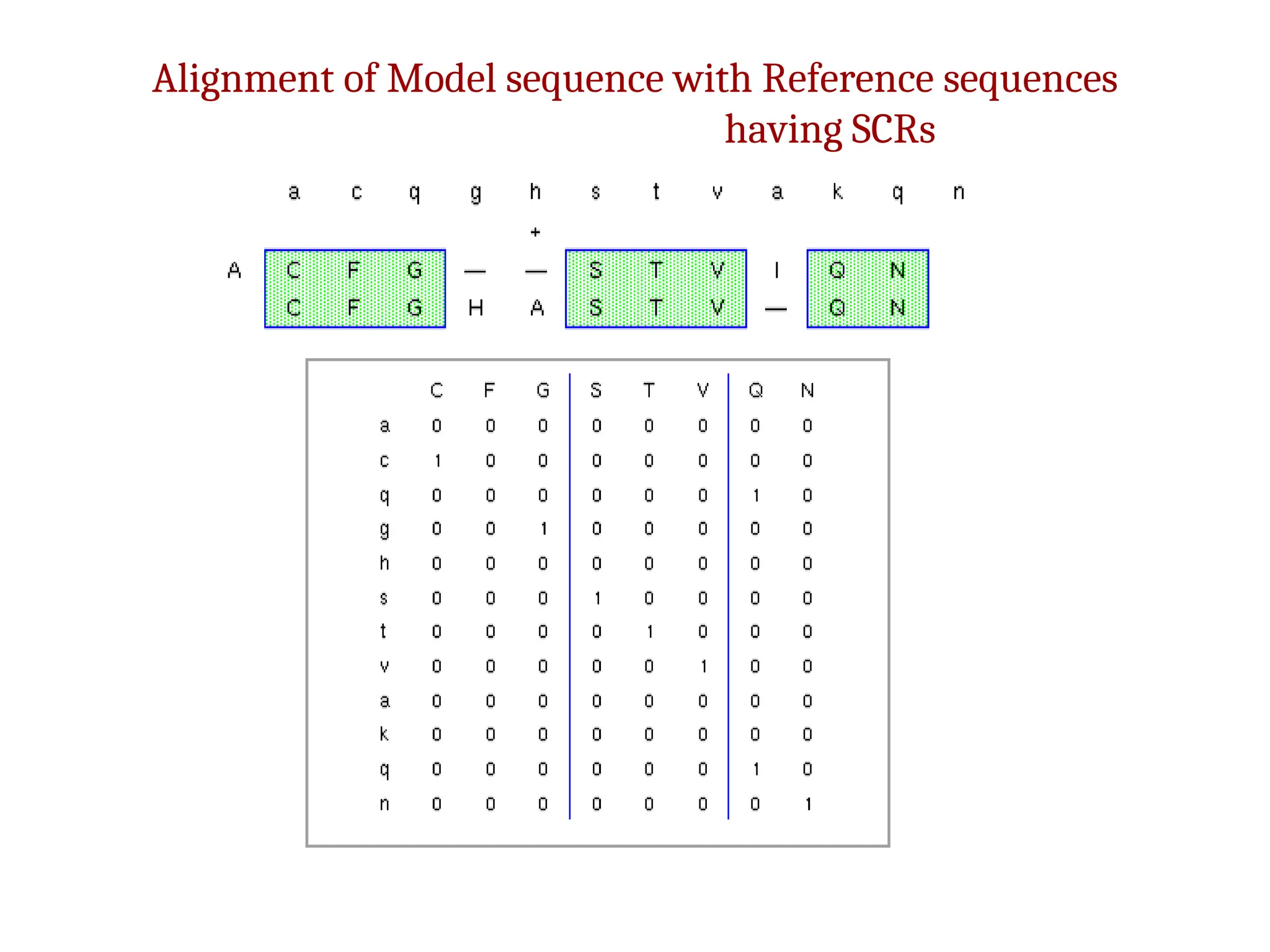
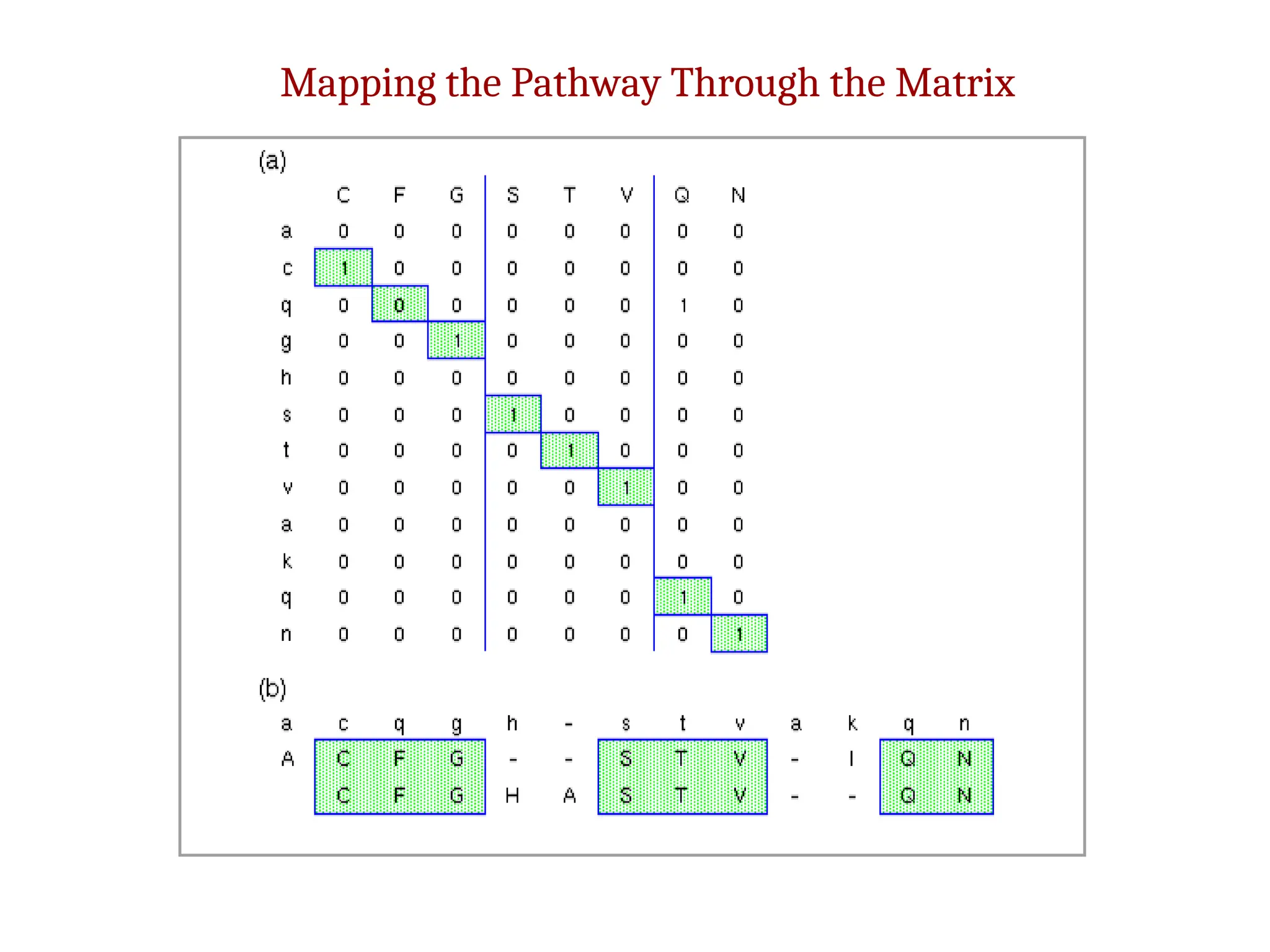
The document discusses homology modeling as a computational method for protein structure prediction, highlighting its reliance on the similarity of amino acid sequences to deduce three-dimensional structures. It outlines the steps involved in homology modeling, including template recognition, sequence alignment, and model refinement, while emphasizing the importance of sequence identity (at least 30%) for constructing reliable models. Additionally, the document mentions various tools and challenges in modeling proteins with lower similarities and the advancements in automated web-based homology modeling.