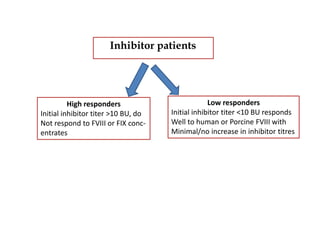
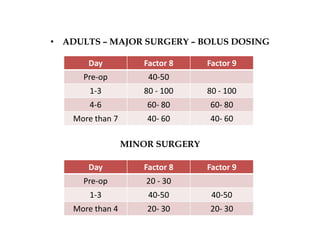
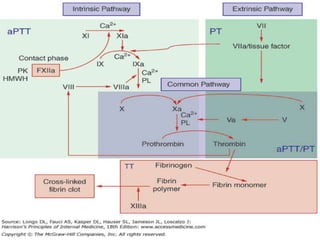
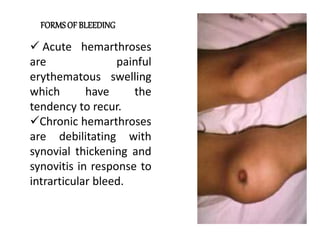
This document provides information on hemophilia, including the objectives, coagulation factors, types and severity of hemophilia, clinical manifestations, complications, labs, treatment including factor replacement therapy and management in special situations. It discusses the basic concepts of hemophilia, how to approach cases, calculate factor requirements, lifestyle modifications, and management during situations like surgery, dental procedures, delivery, and menstruation in hemophiliacs.


















![NON TRANSFUSION THERAPHY IN
HEMOPHILIA
Mild-to-moderate hemophilia A with minor bleeding:
* DDAVP (0.3 mg/kg IV in 50 to 100 mL NS infused over 30 minutes, or 300
mg intranasally [Stimate, 1.5 mg/mL] dosed every 12 hours). Increases factor VIII
activity threefold to fivefold and has a half-life of 8 to 12 hours. Tachyphylaxis
may occur after several doses
•ANTI-FIBRINOLYTIC AGENTS: EACA or Tranexemic acid.
Bleeding in gums, GIT or during oral surgery. EACA – loading dose of
200mg/kg followed by 100 mg/kg per dose every 6th hourly. Tranexemic Acid – 25 mg/kg 3
to 5 times a day.
Not indicated to control hematuria because of the risk of formation of occlusive clot in the
lumen of genitoruinary tract structues](https://image.slidesharecdn.com/hemophila-190121092014/85/Hemophila-27-320.jpg)