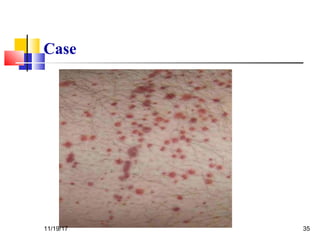
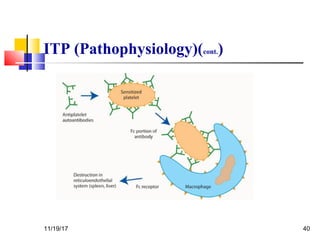
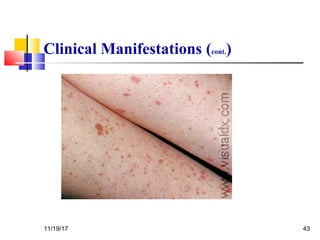
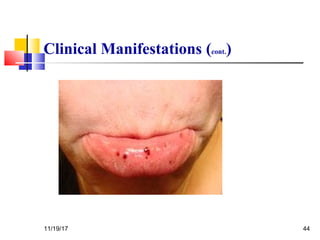
A 3 year old female child presented with red spots on her lower limbs that appeared in the morning. She had a history of an upper respiratory infection around 3 weeks prior. On examination she was normal. Her platelet count was 20k and other blood tests including hemoglobin were normal. This presentation is suggestive of idiopathic thrombocytopenic purpura (ITP), the most common cause of acute onset thrombocytopenia in a previously well child. ITP often occurs 1-4 weeks after a viral infection, with the peak age being 1-4 years old. The pathophysiology involves an autoantibody developing against platelets, leading to their destruction in the spleen.















































































































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)










