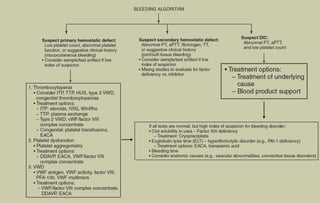
The document provides a comprehensive overview of bleeding disorders in children, focusing on diagnostic tests for platelet and coagulation factor disorders, including conditions such as hemophilia and von Willebrand disease (vWD). It details clinical manifestations, investigation methods, and management protocols including treatment options like desmopressin and factor concentrates. The document emphasizes the importance of early diagnosis and appropriate therapy to manage bleeding tendencies effectively.
























































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)









![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)





