








![DEPENDING ON CHARACTERS
TWO TYPES OF THALASSAEMIA
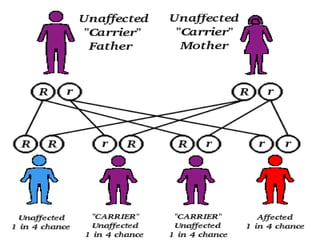
HETEROZYGOUS STATE [α and β thalassaemia
minor or trait]- generally asymptomatic
HOMOZYGOUS STATE [α and β thalassaemia
major]-congenital haemolytic](https://image.slidesharecdn.com/thalassaemias-sarasjothi-121231075103-phpapp01/85/Thalassaemias-By-Sarasjothi-13-320.jpg)




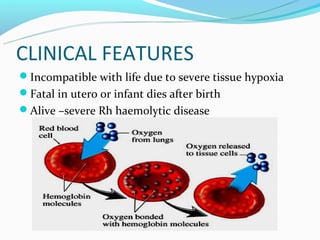


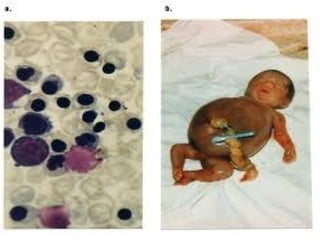
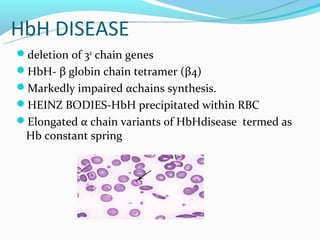

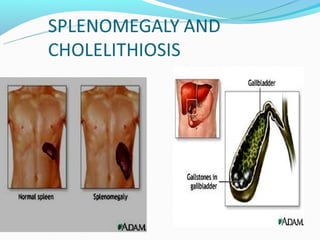

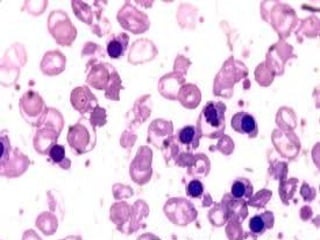



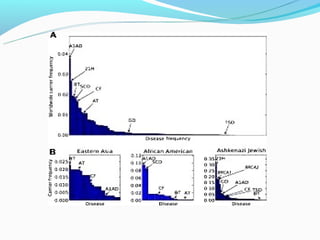

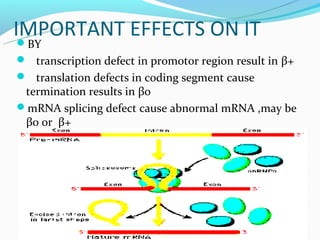
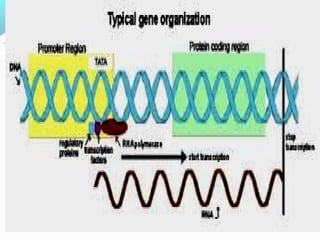



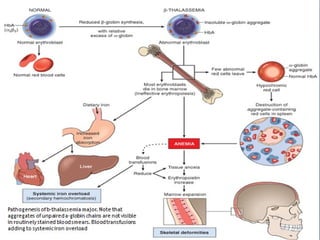

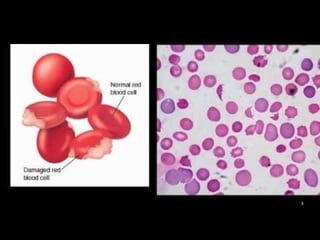


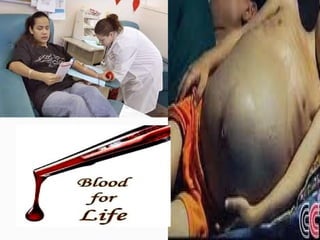


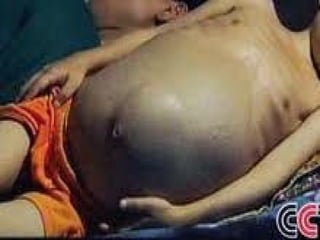

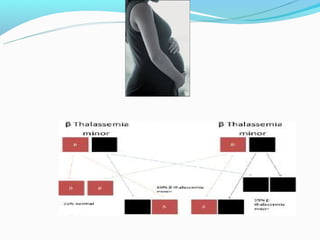


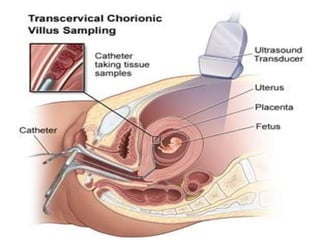




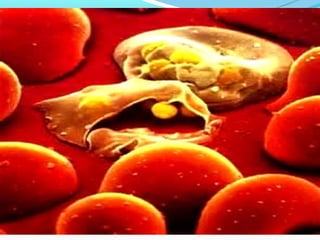
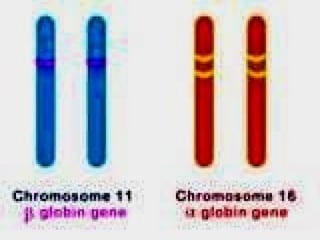
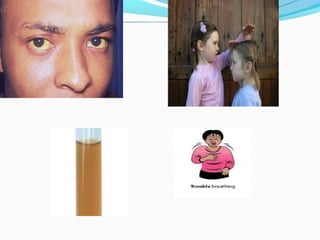
This document discusses thalassemias, which are hereditary disorders involving reduced synthesis of one or more globin polypeptide chains. It is classified into alpha and beta thalassemias. Alpha thalassemia involves defective synthesis of alpha globin chains, while beta thalassemia involves reduced beta chain synthesis. The classification ranges from severe homozygous forms like hydrops fetalis to mild heterozygous forms like thalassemia minor. The clinical features, laboratory findings, and treatment approaches are described for different forms of alpha and beta thalassemias. Prevention can be achieved through antenatal screening and diagnosis.





































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)








