Downloaded 82 times





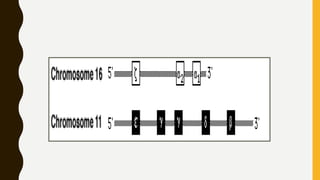
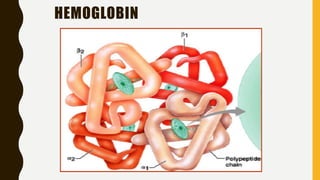
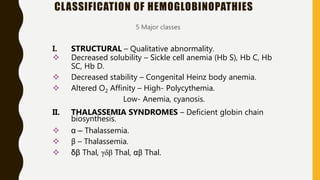








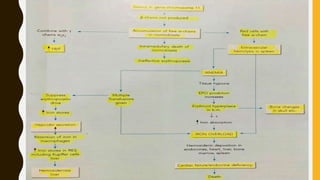
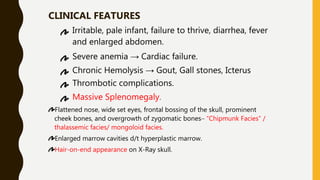
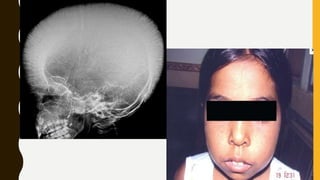

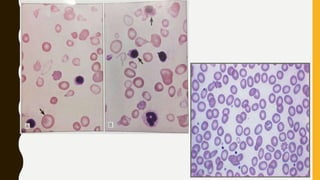


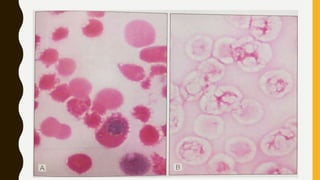

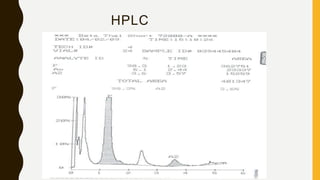
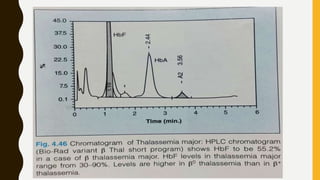
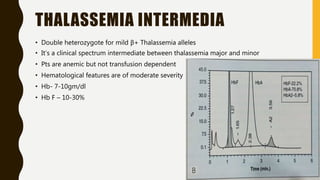

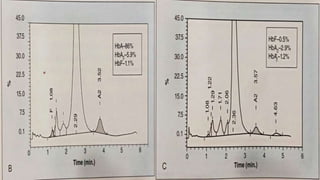

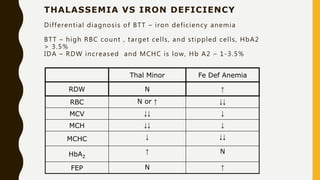







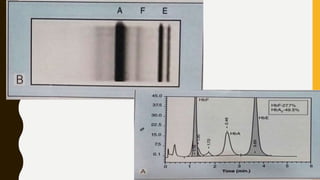

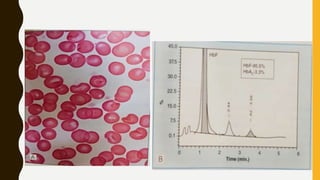



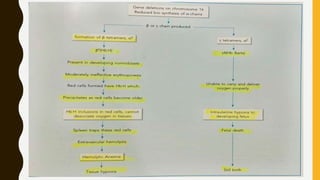

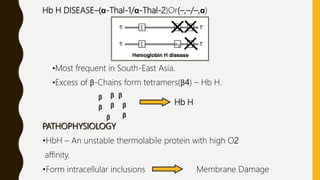

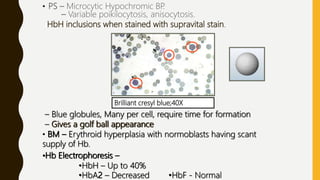





This document provides information on hemoglobinopathies and thalassemias. It begins by defining hemoglobinopathies as genetically determined abnormalities of the hemoglobin molecule, associated with globin chains. It then discusses the different types of globin chains and normal hemoglobin development. The document classifies hemoglobinopathies into 5 major classes including structural abnormalities, thalassemia syndromes, thalassemic hemoglobin variants, hereditary persistence of fetal hemoglobin, and acquired hemoglobinopathies. It provides detailed information on beta thalassemia syndromes, genetics, clinical features, hematologic findings, bone marrow findings, laboratory tests, and complications/management of thalassemia major. It also briefly discusses





































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)



![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)

