Download as PDF, PPTX












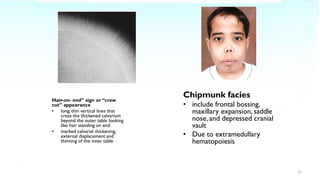





Thalassemia is a blood disorder caused by reduced or absent globin chain production, leading to anemia. It is classified as alpha or beta thalassemia depending on the deficient chain. Common in areas like Southeast Asia and the Mediterranean. Diagnosis involves blood tests showing hypochromic microcytic anemia and globin gene testing. Symptoms range from mild to severe depending on the number of defective genes. The most severe form is beta thalassemia major requiring lifelong blood transfusions and iron chelation therapy. Complications include anemia, jaundice, bone changes, iron overload affecting organs, and growth issues.






































![THALASEMIA definition and pathophysiologyd].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/thalasemia-240515044127-8ce804c5-thumbnail.jpg?width=640&height=640&fit=bounds)

































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)
![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)











