Downloaded 15 times






















































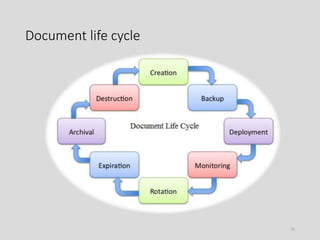




The document discusses the importance of documentation in quality assurance for the pharmaceutical industry, outlining its role in compliance with regulations, quality control, and operational efficiency. It highlights the need for clear and systematic documentation practices, including the design, approval, review, and archiving of various types of records related to drug manufacturing. Essential features of good documentation are also emphasized, such as accuracy, clarity, and regular updates to ensure adherence to legal and quality standards.








































![supriya.k_ppt[1].pptx documentation for QA and QC](https://cdn.slidesharecdn.com/ss_thumbnails/supriya-250809112814-eb4e6f1f-thumbnail.jpg?width=640&height=640&fit=bounds)














































