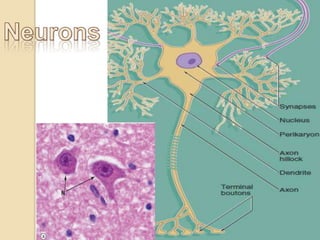
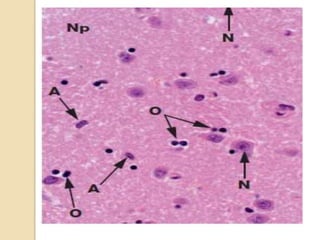
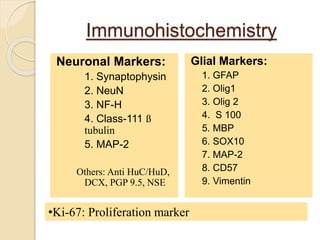
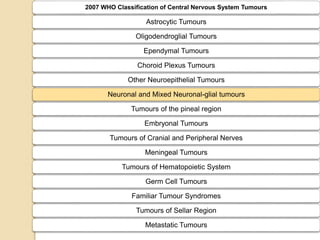
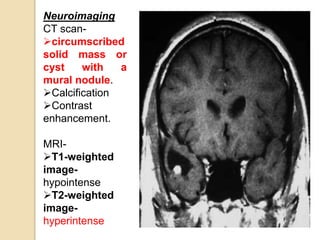
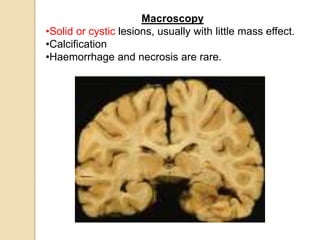
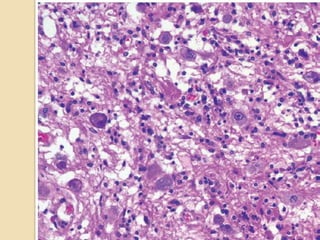
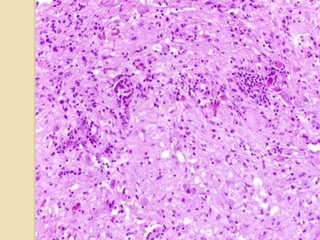
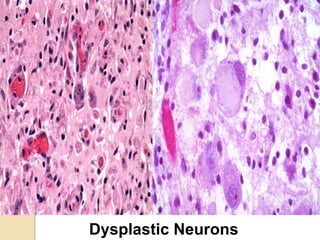
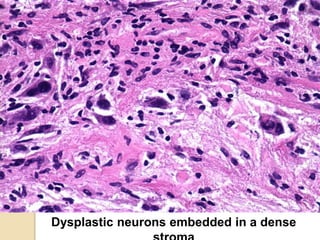
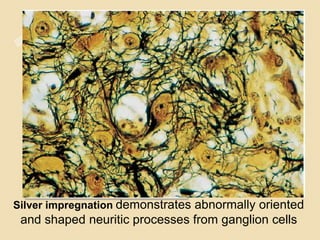
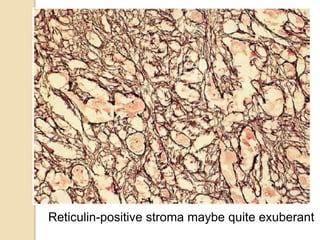
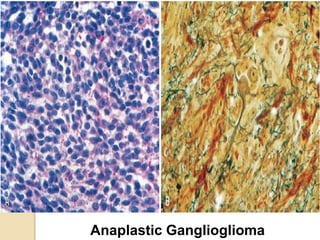
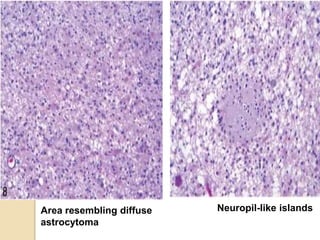
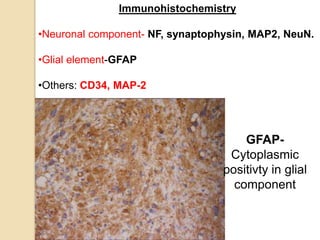
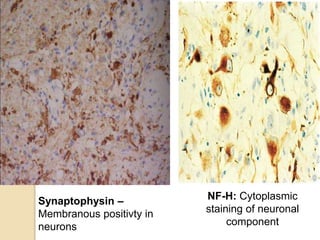
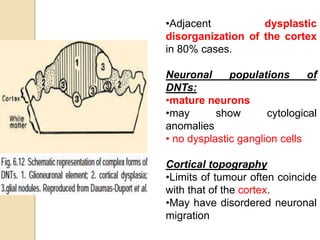
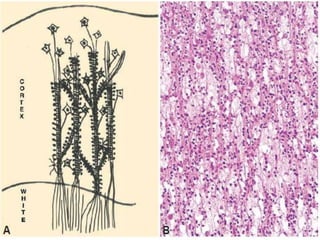
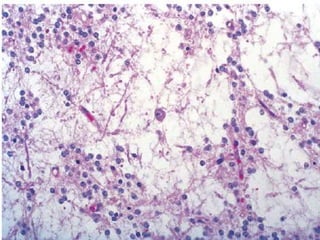
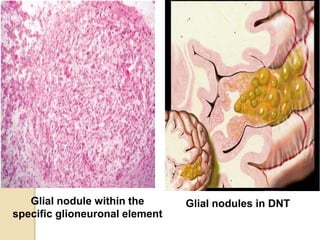
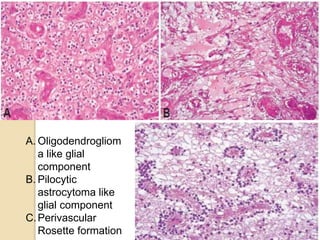
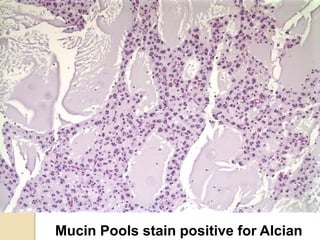
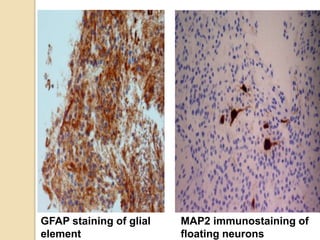
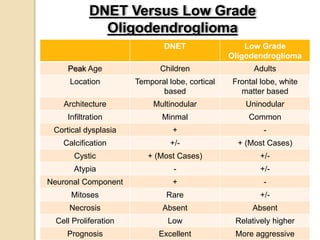
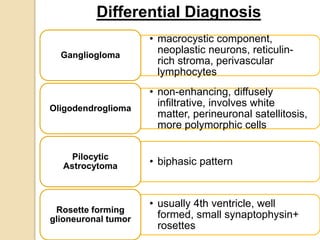
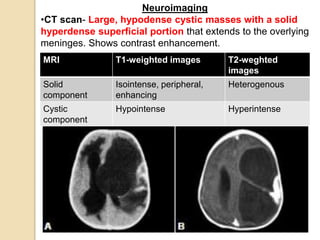
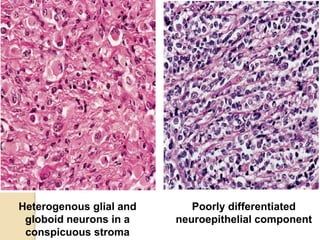
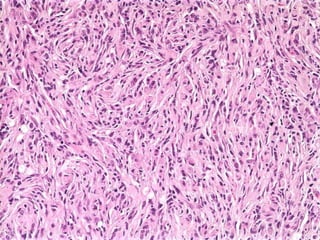
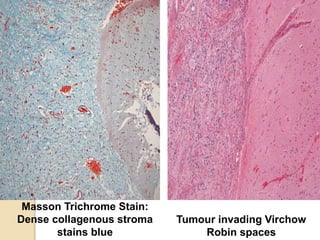
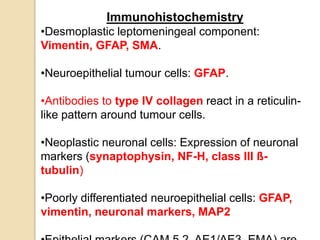
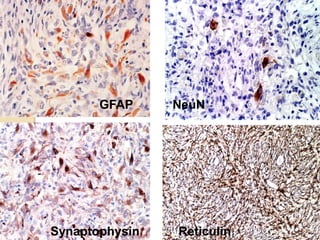
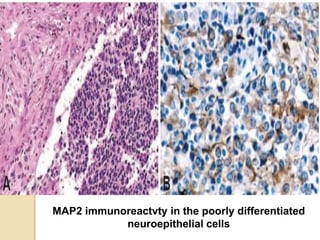
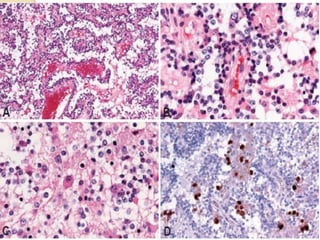
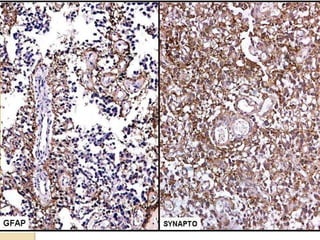
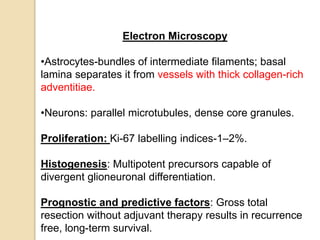
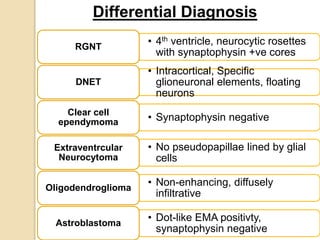
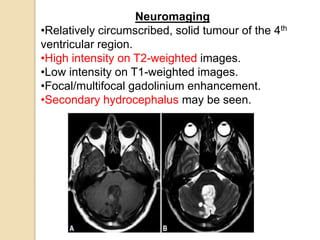
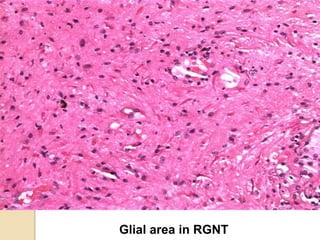
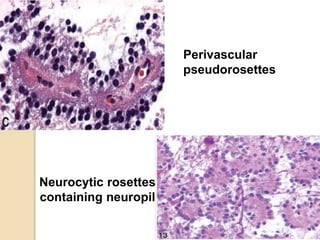
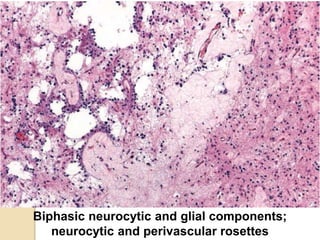
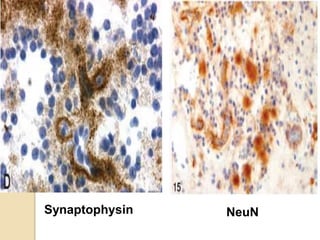
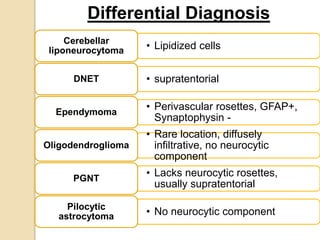
The document discusses glioneuronal tumors, outlining their types, characteristics, and classification according to the 2007 WHO guidelines. It explains the clinical significance of these tumors, emphasizing their favorable outcomes and treatment options compared to gliomas. Specific types such as gangliogliomas and dysembryoplastic neuroepithelial tumors are detailed with their incidence, features, and differential diagnoses.