Downloaded 29 times







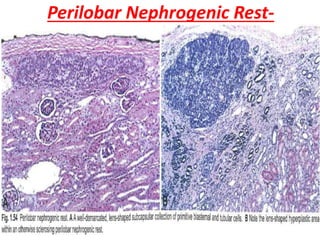
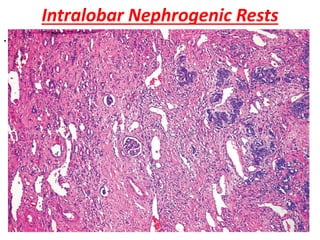


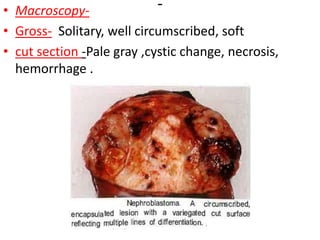
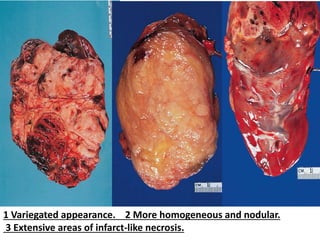

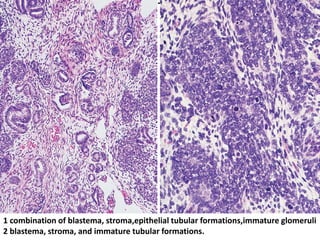
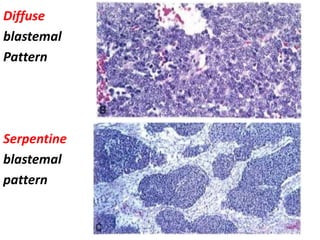
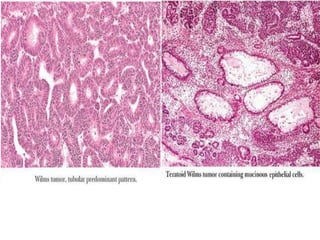





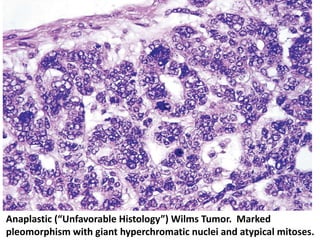




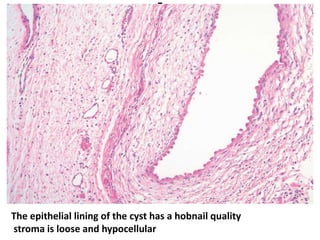



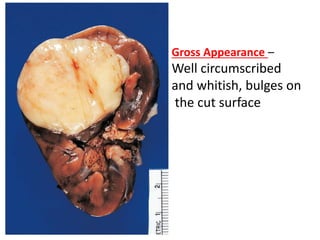

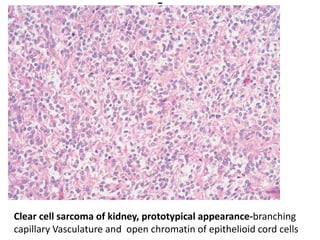
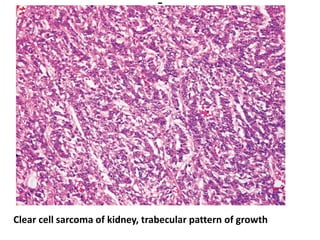
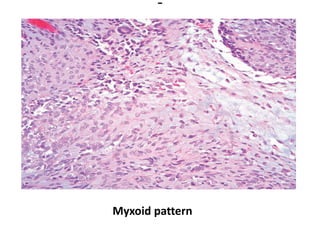
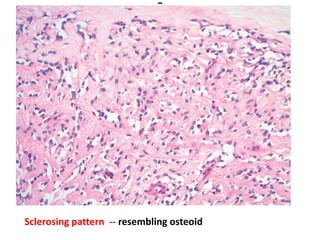
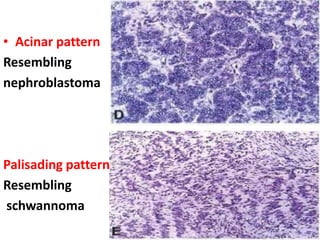
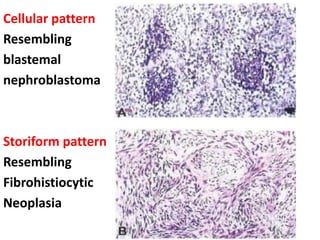





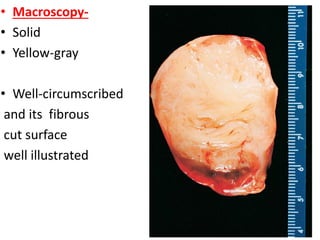
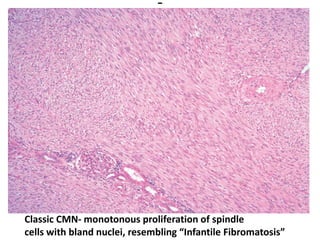
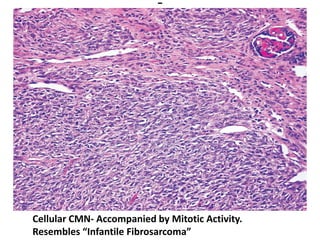



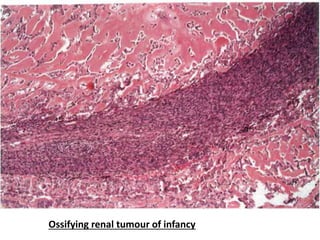



The document discusses pediatric renal tumors, categorizing them into nephroblastic and cystic tumors as well as mesenchymal tumors, detailing specific types such as Wilms tumor, nephrogenic rest, and others. It includes information on epidemiology, clinical features, histopathology, molecular genetics, and treatment protocols for various tumor types. Prognostic factors and associated syndromes are also highlighted, providing a comprehensive overview of the pathology related to pediatric renal tumors.

































![Wilm's tumour - The most common kidney tumor in children - Dr Vishnu A [VCR],...](https://cdn.slidesharecdn.com/ss_thumbnails/vishnu-wilmstumour-210312145616-thumbnail.jpg?width=640&height=640&fit=bounds)

































