Downloaded 620 times





















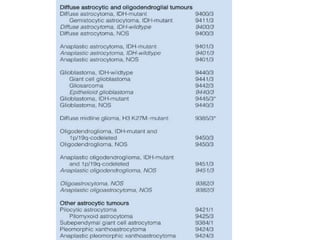



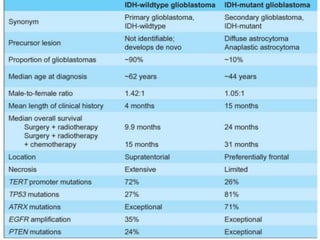


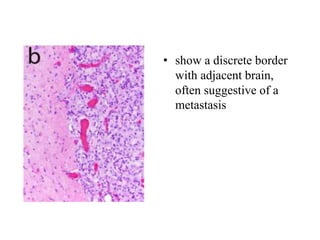
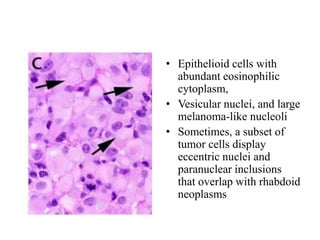
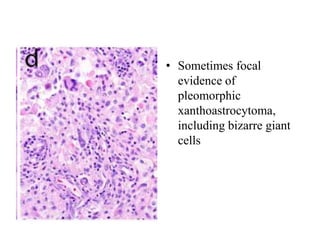
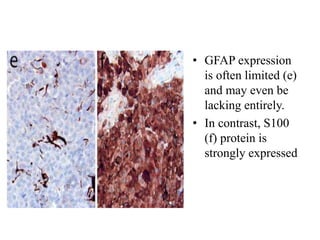
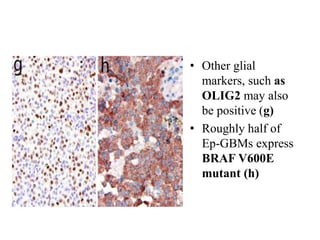


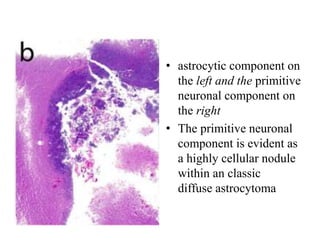
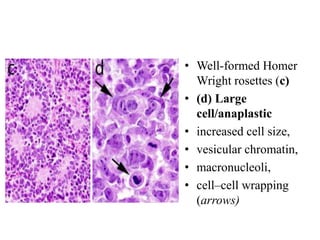
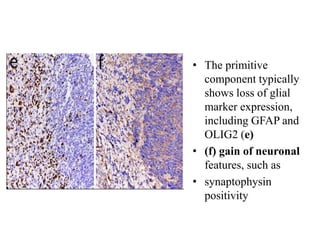
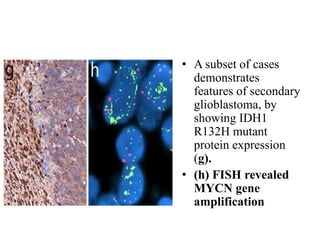




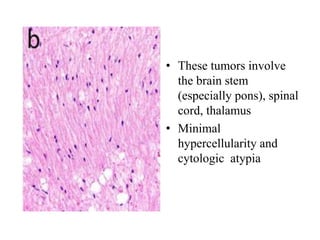
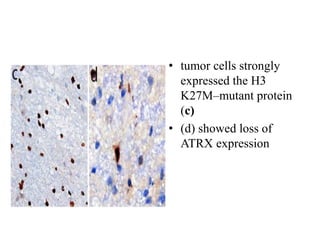
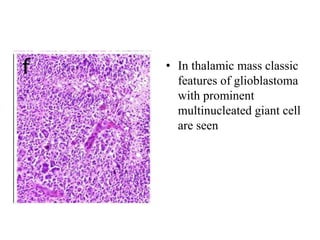
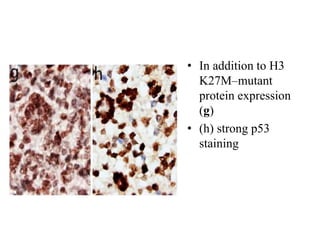


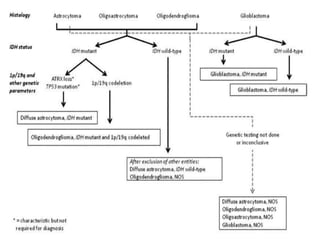

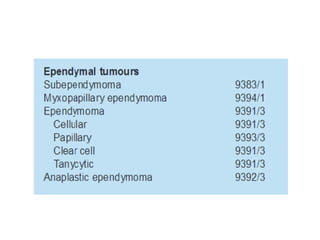
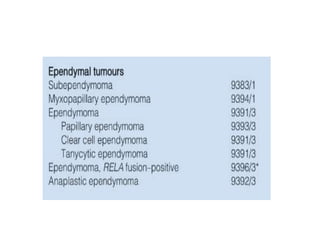


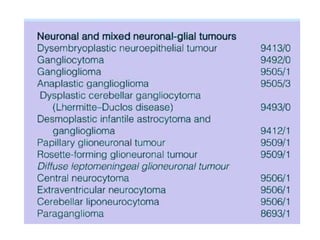

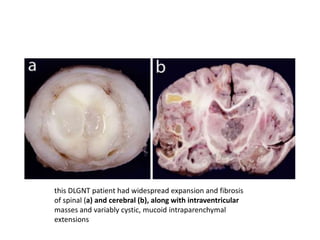
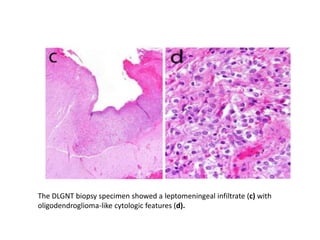
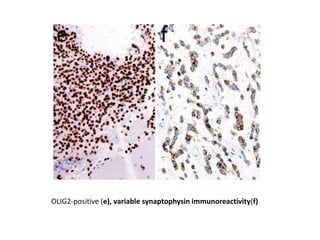
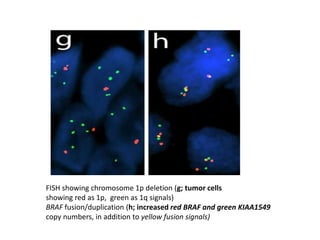

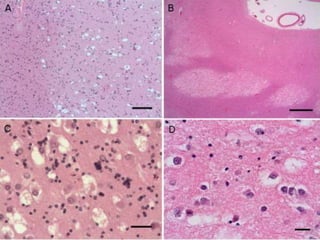



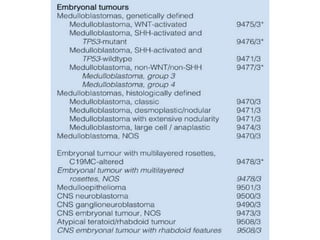
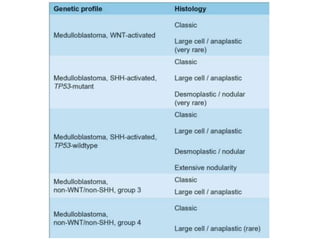






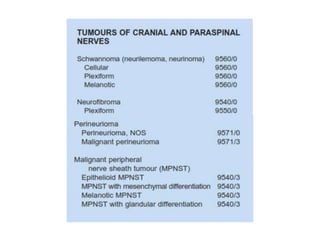
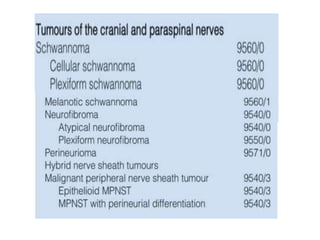






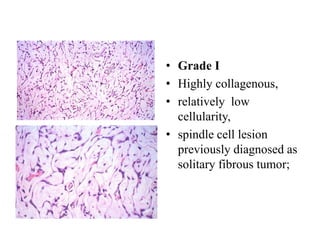
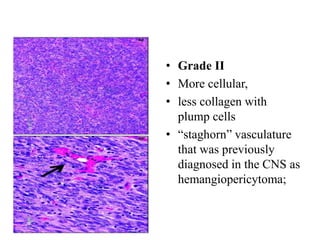
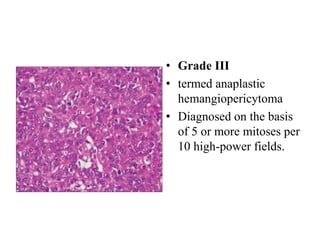


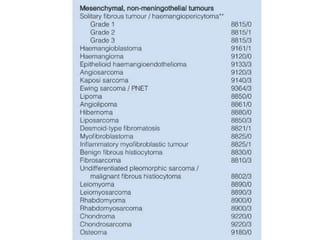




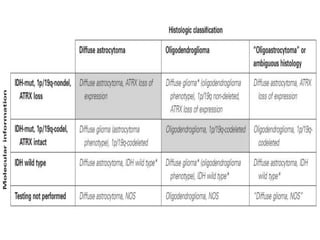











This document provides an overview of recent updates to the WHO classification of central nervous system tumors based on the 2016 guidelines. Key points include: - Incorporation of molecular parameters like IDH, ATRX, and 1p/19q status into tumor classifications to improve diagnostic accuracy. - Diffuse gliomas are now classified based on shared genetic drivers rather than histology alone. Entities like oligoastrocytoma are discouraged. - Newly recognized entities include epithelioid glioblastoma and glioblastoma with a primitive neuronal component showing MYC/MYCN amplification. - The diagnosis of oligodendroglioma now requires both IDH mutation and 1p/










































































