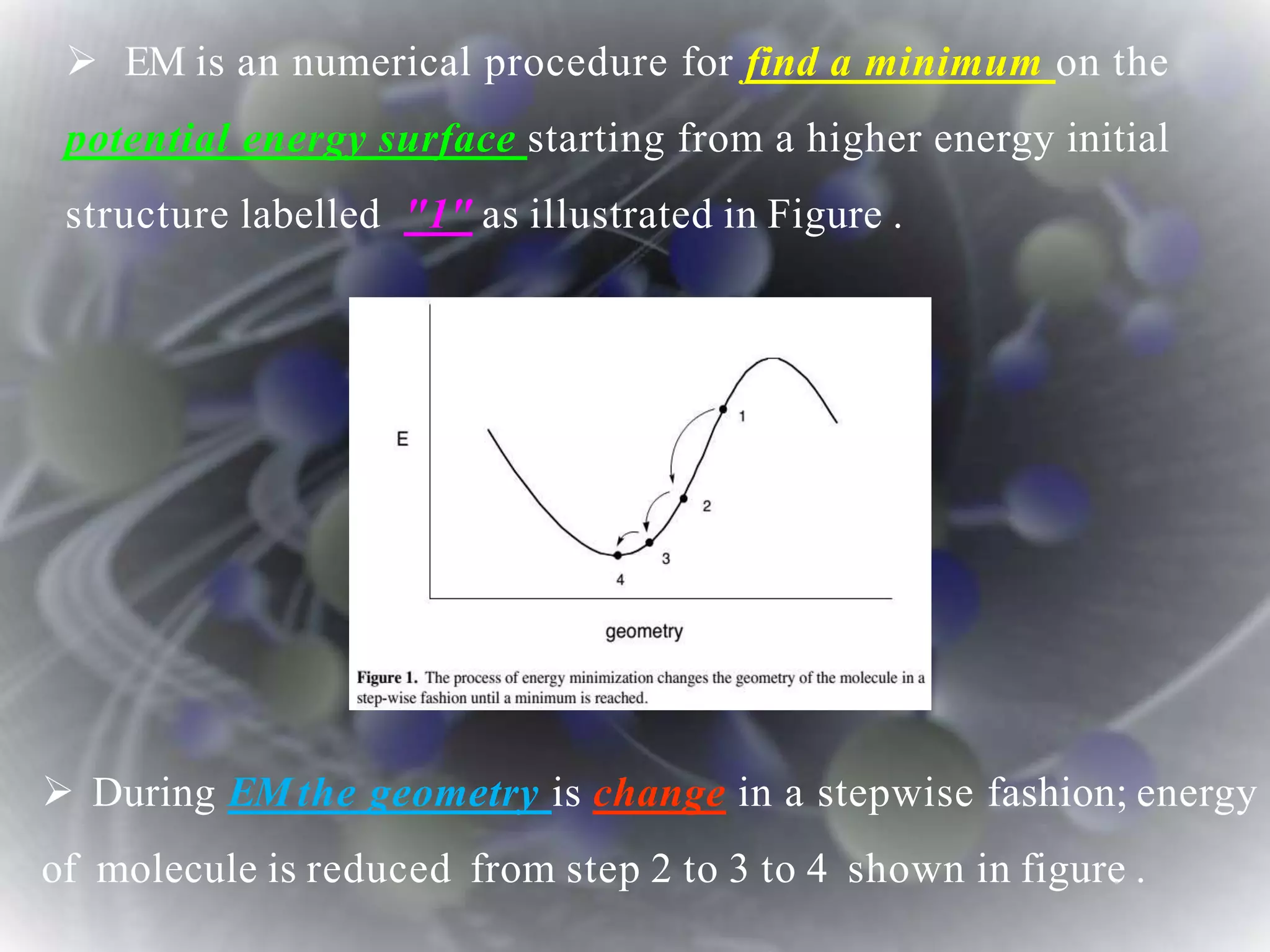
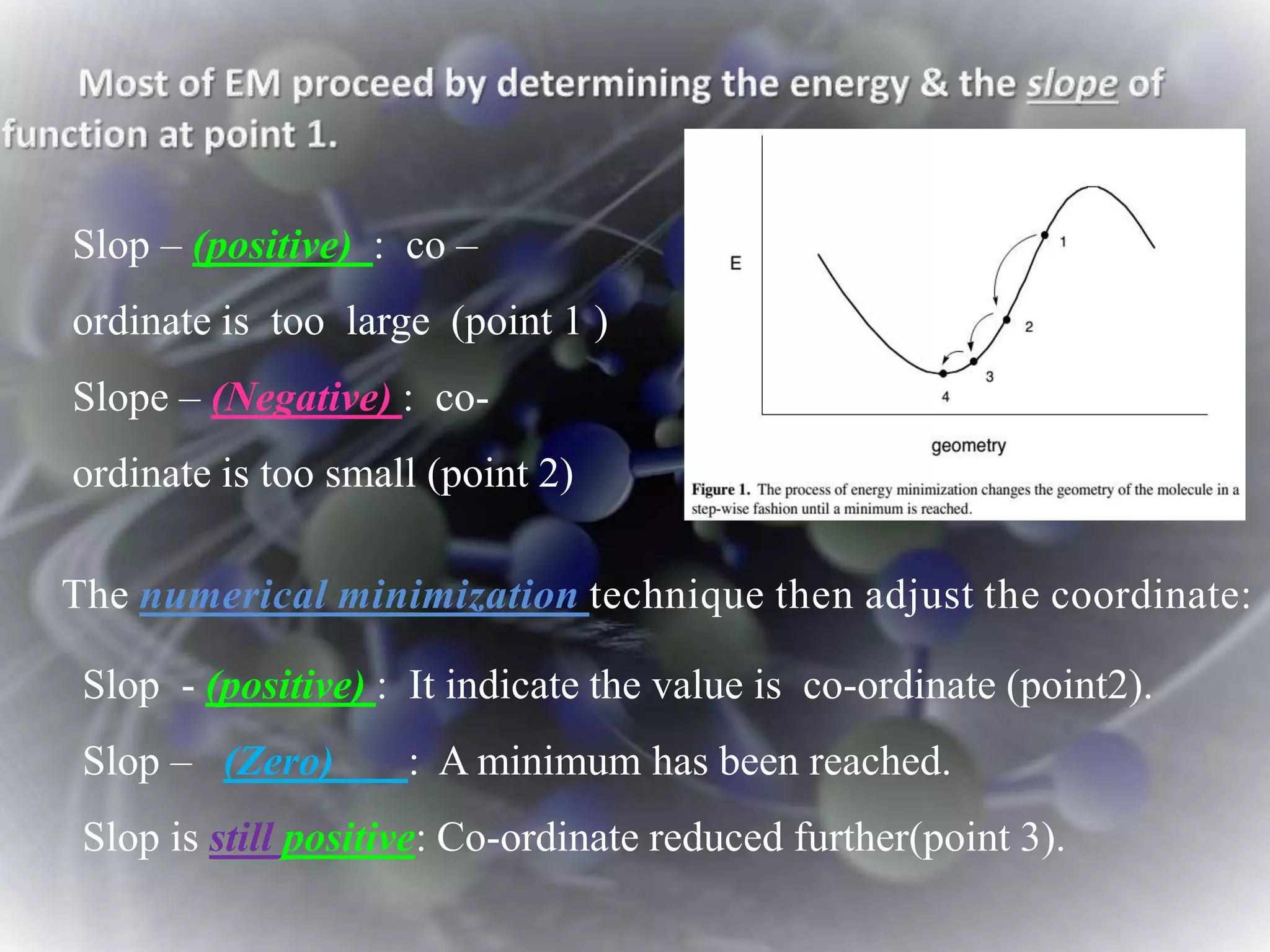
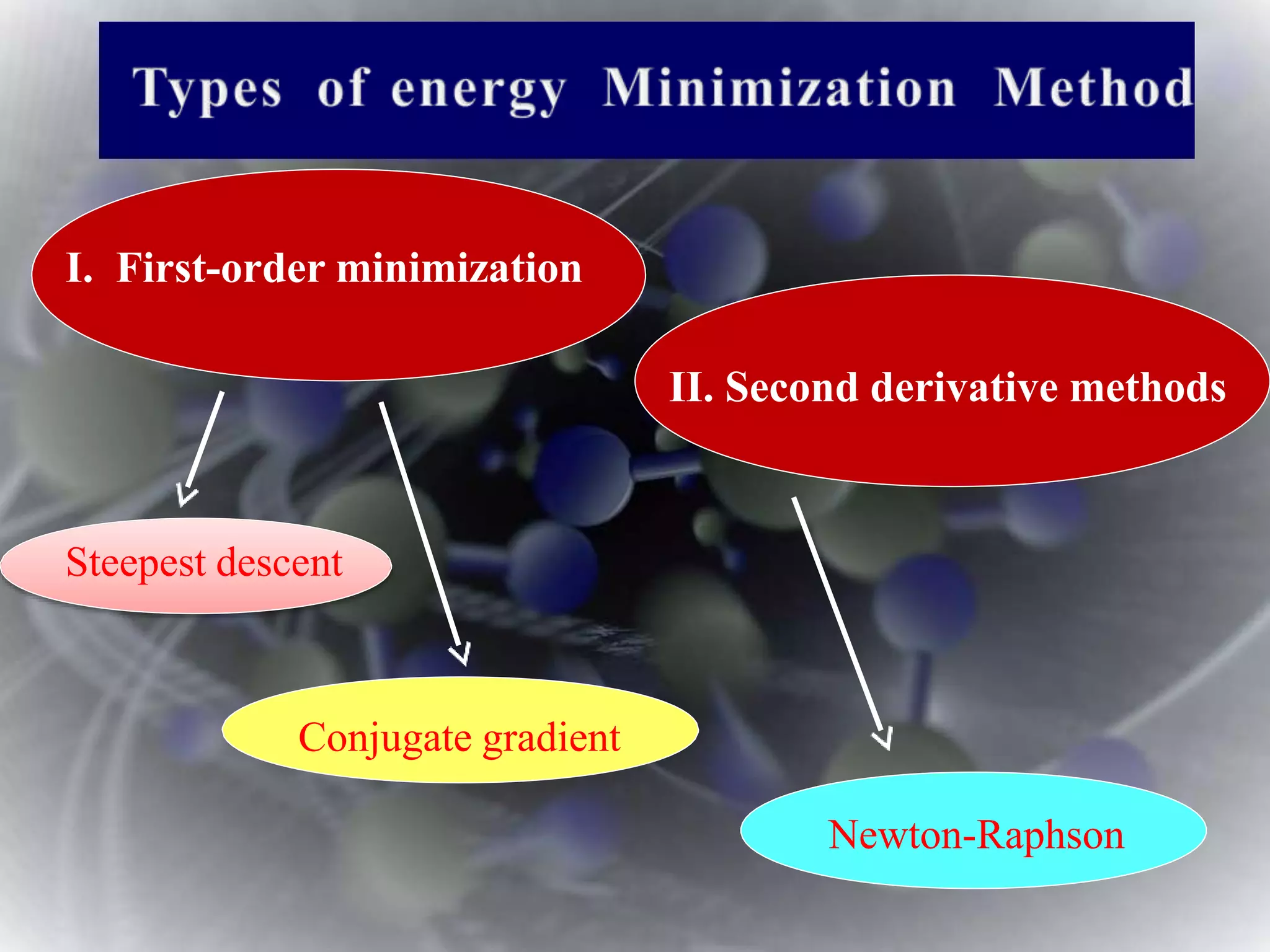
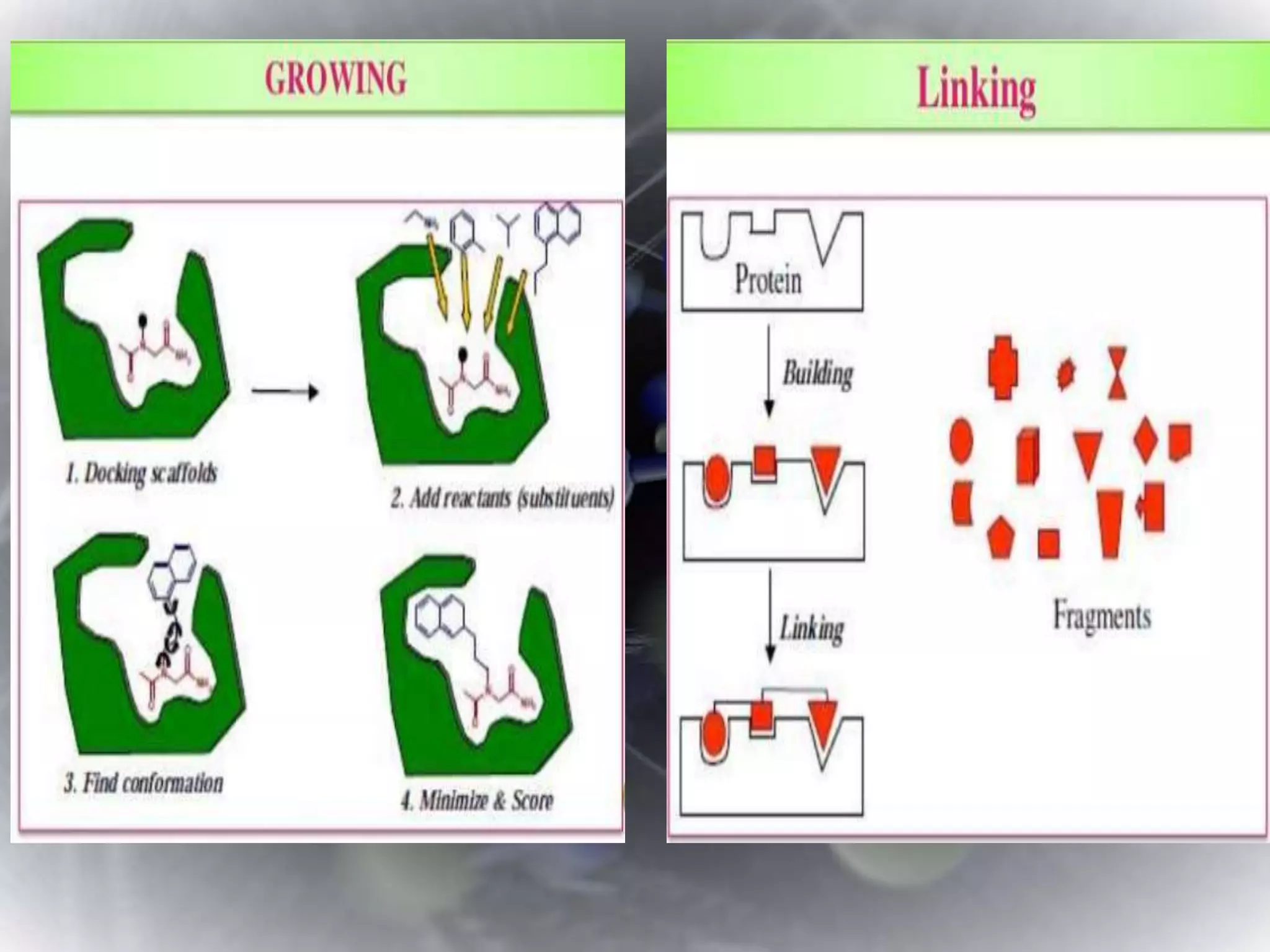
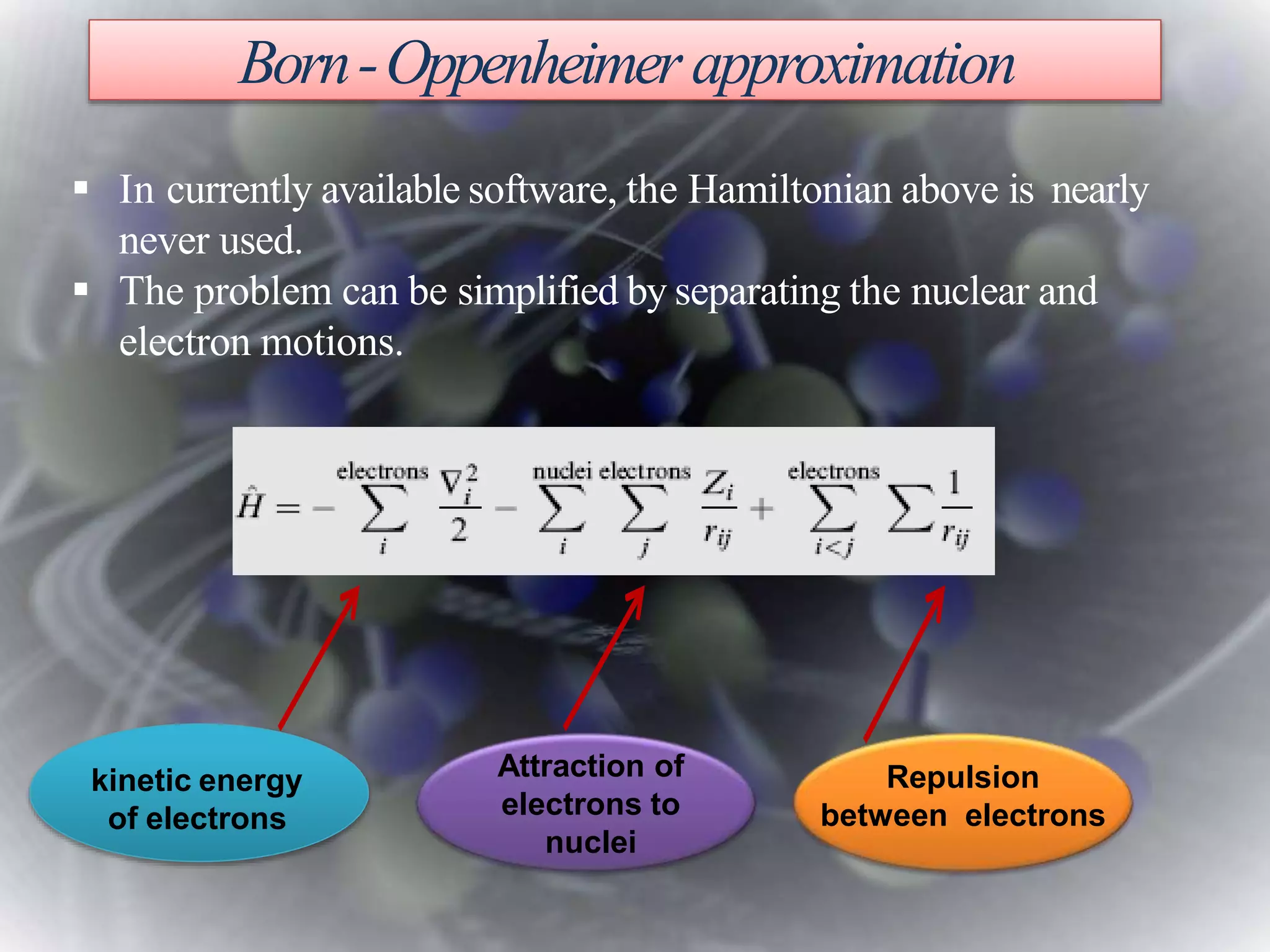
The document discusses various molecular modeling and computational chemistry techniques used to simulate molecular systems, including molecular dynamics, molecular mechanics, quantum mechanics methods, and molecular docking. It provides an overview of the different modeling strategies and computational tools used, such as determining receptor geometry from X-ray crystallography, energy minimization techniques, force field parameters, and quantum mechanical calculations. The goal of molecular modeling is to develop accurate models of molecular systems to predict properties and behavior without experimental testing.
















![Force field:
It is set of function & constant used to described potential energy of
the molecule.
General form of force field equation ;
Epot = ∑Ebon+ ∑Eang+ ∑Etor + ∑Eoop+ ∑ Enb + ∑Eel
Where;
Epot = The total steric energy
Ebon = The energy resulting from changing the bond
length from it’s initial value calculated by Hook’s law for
deformation spring E=1/2kb(b-b0)2
[ kb = force constant for bond, b0-equilibrium bond length ,b-current
bond length]
Eang = The energy resulting from deforming a bond angle
from it’s original value .
Etor : Deforming the torsinal or dihydral angle
Eoop : Is the out of plane bending component of the steric
energy](https://image.slidesharecdn.com/molecularmodellinganddocking-iyr-2-1s-190326091622/75/Molecular-modelling-and-docking-studies-18-2048.jpg)