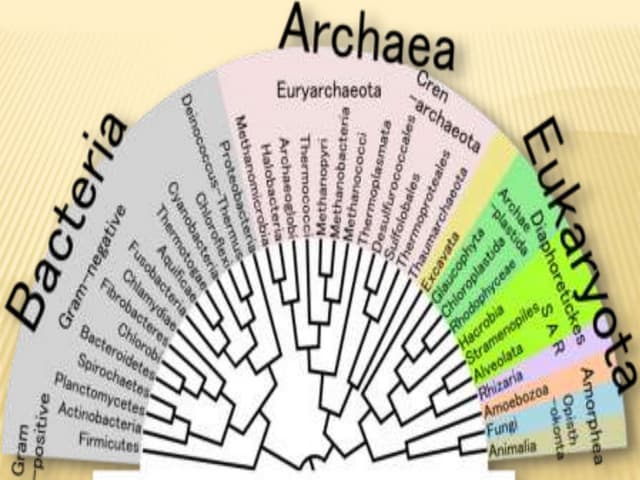
UPGMA (Unweighted Pair Group Method with Arithmetic Mean) is a distance-based method for constructing phylogenetic trees, developed in 1958 by Sokal and Michener. It employs a sequential clustering algorithm using a distance matrix to generate rooted, ultra-metric trees, reflecting phenotypic similarities. While simple and fast, UPGMA has limitations, such as assuming constant evolutionary speed across lineages, which can lead to inaccurate tree topologies.