Downloaded 114 times



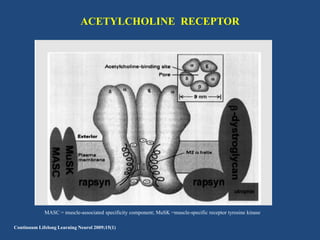



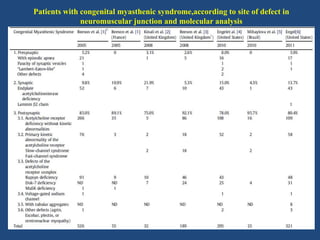

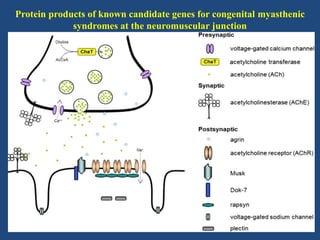
















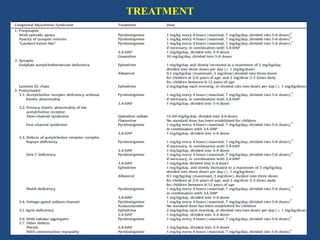



This document discusses congenital myasthenic syndromes (CMS), which are rare genetic disorders characterized by compromised neuromuscular transmission. CMS are classified based on the location of the primary defect within the neuromuscular junction, including presynaptic, synaptic, and postsynaptic defects. The majority of cases involve postsynaptic defects. Over 20 genes have been identified that can cause CMS. Clinical features typically include fatigable weakness since infancy and decremental responses on electrodiagnostic testing. Treatment involves cholinesterase inhibitors and other medications depending on the specific CMS subtype. Differential diagnosis includes other neuromuscular disorders presenting in infancy. Definitive diagnosis may require genetic and other specialized testing.















































![NEUROMUSCULAR JUNCTION [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/neuromuscularjunctionautosaved-230730051121-668f660d-thumbnail.jpg?width=640&height=640&fit=bounds)





















![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)
















