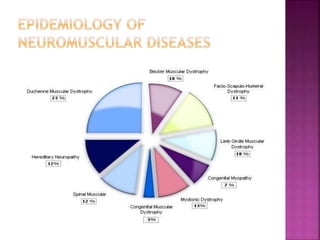
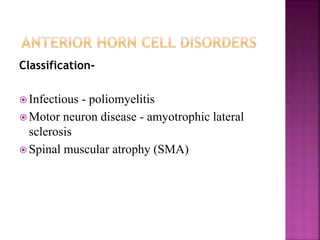
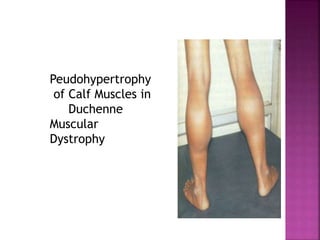
The document discusses neuromuscular diseases. It defines the motor unit and its components. Some key neuromuscular diseases discussed include muscular dystrophies, congenital and metabolic myopathies, anterior horn cell disorders, and neuromuscular junction diseases. Common symptoms of neuromuscular diseases are then outlined. Diagnostic tests and treatment approaches are also summarized for several specific conditions like spinal muscular atrophy, Guillain-Barré syndrome, and myasthenia gravis.