Downloaded 242 times









































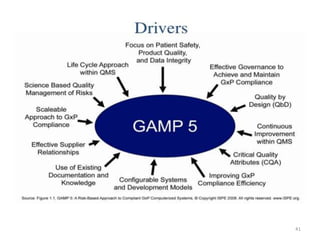
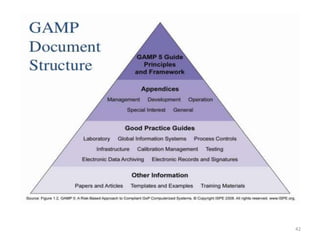


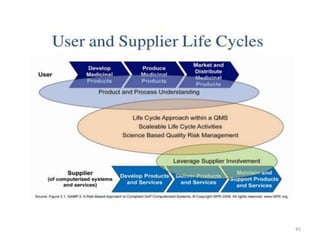


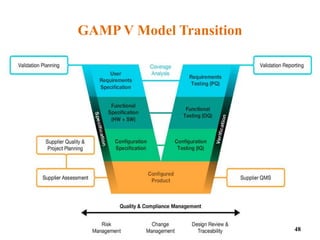



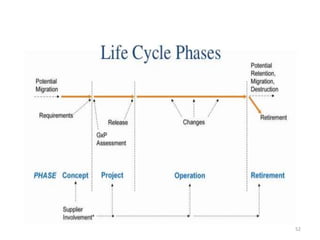

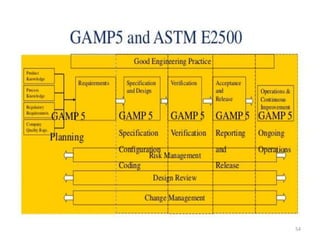
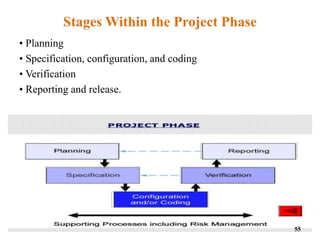


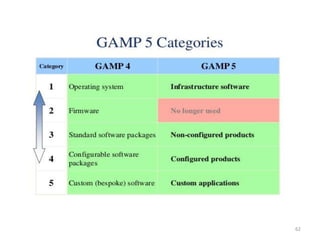


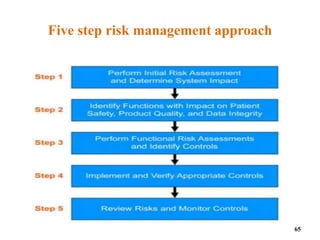



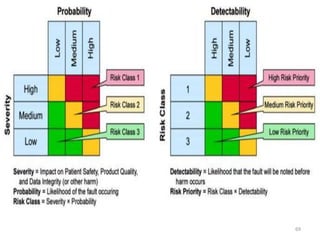

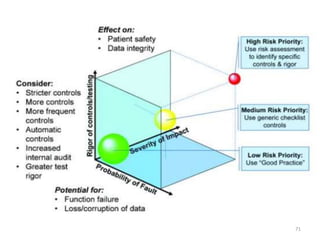





This document discusses computer system validation and 21 CFR Part 11 requirements for electronic records and signatures. It provides definitions of key terms from Part 11 such as electronic record, electronic signature, closed system and open system. It also summarizes the controls required by Part 11 for closed and open systems, including validation, audit trails, limiting access, documentation controls and more. The purpose is to ensure electronic records and signatures are trustworthy and equivalent to paper records.















































































