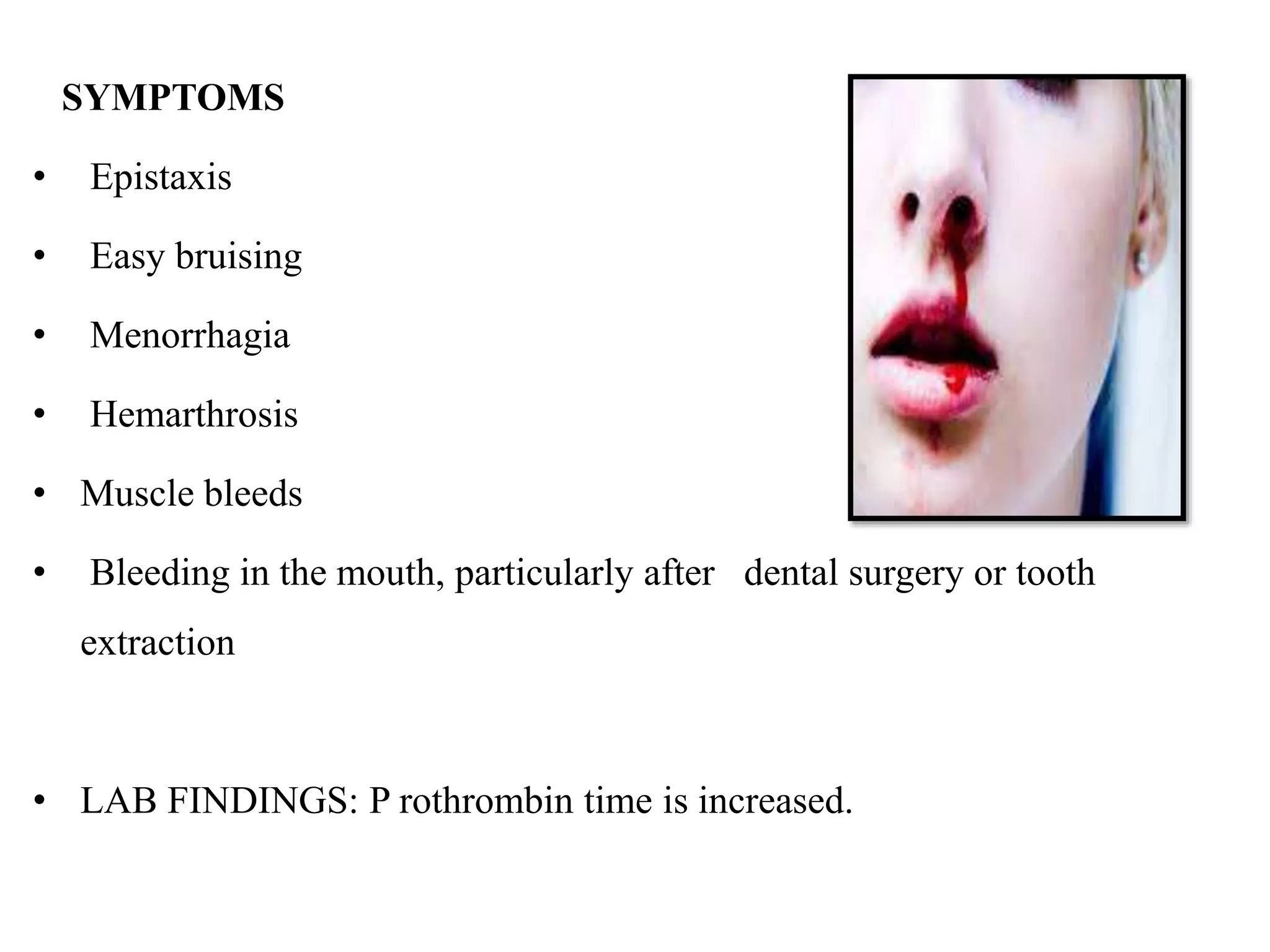
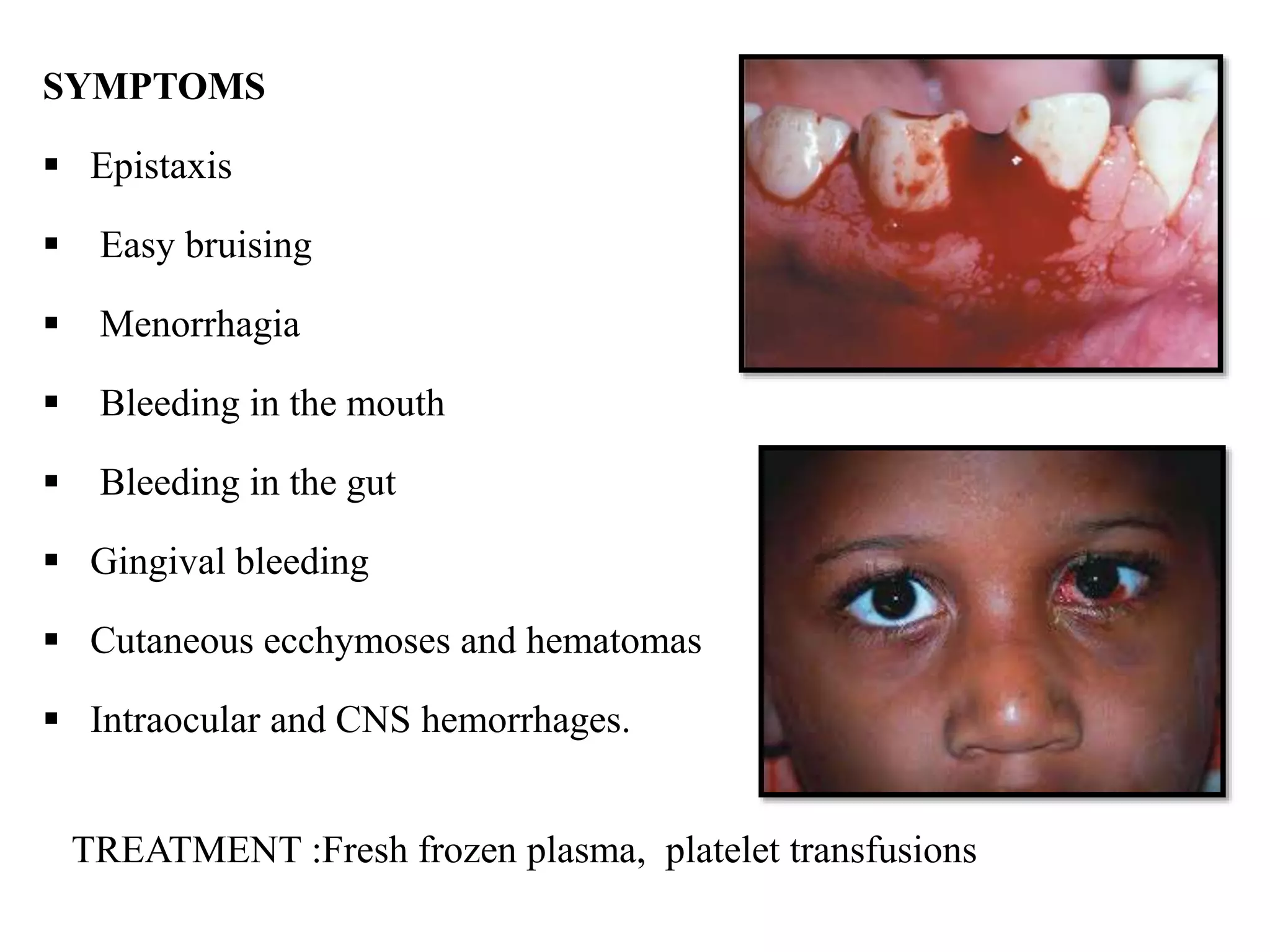
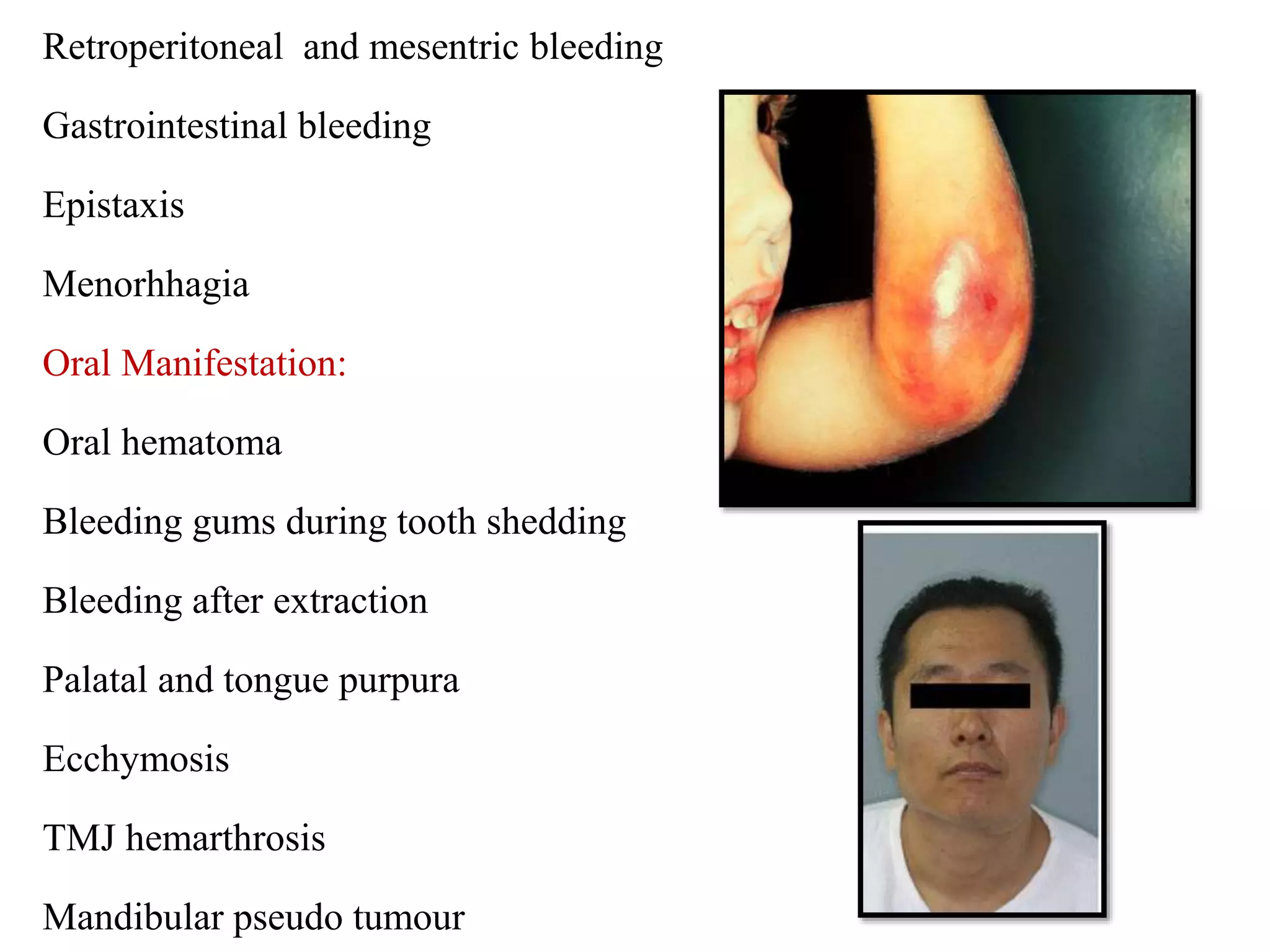
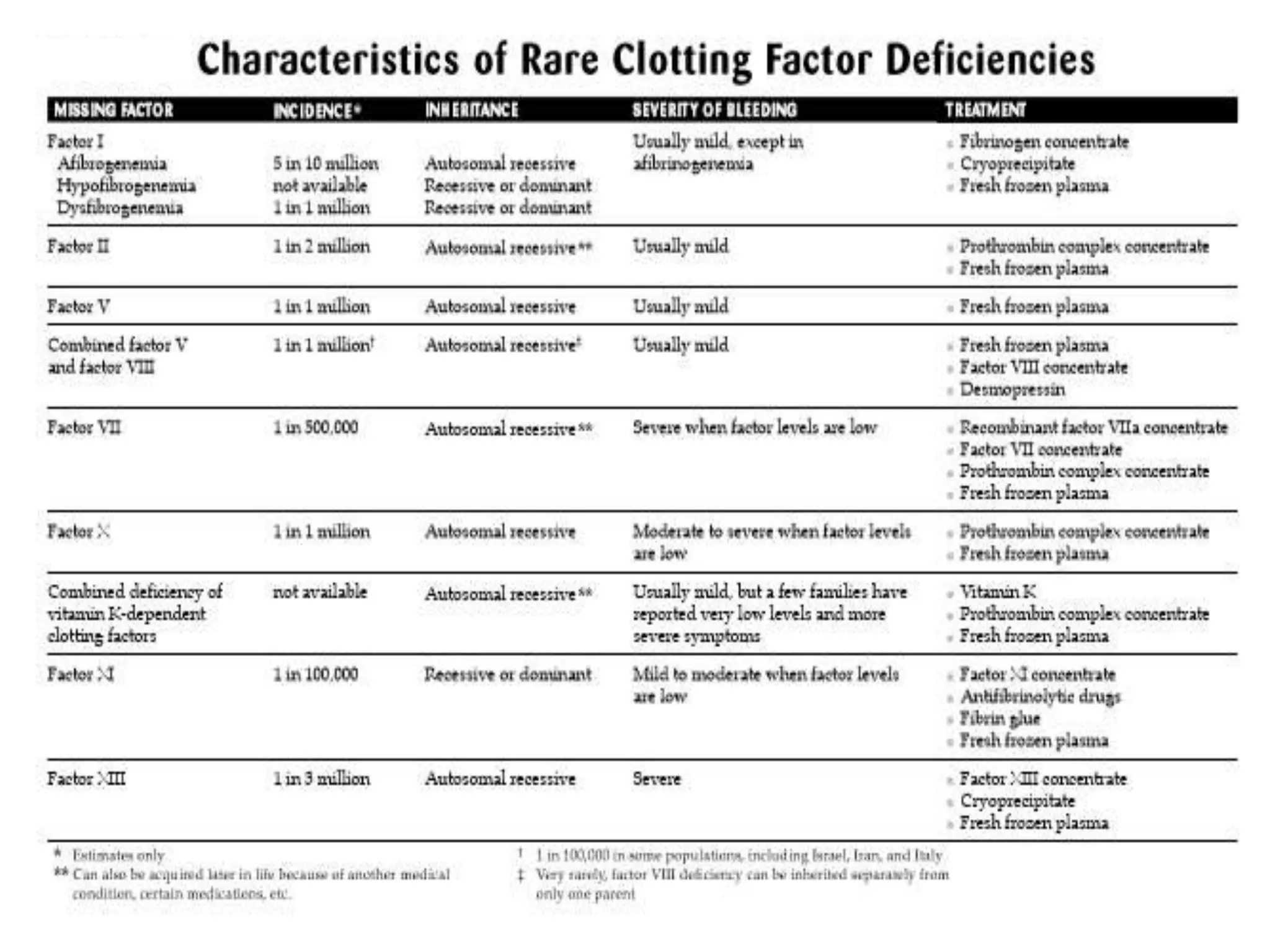
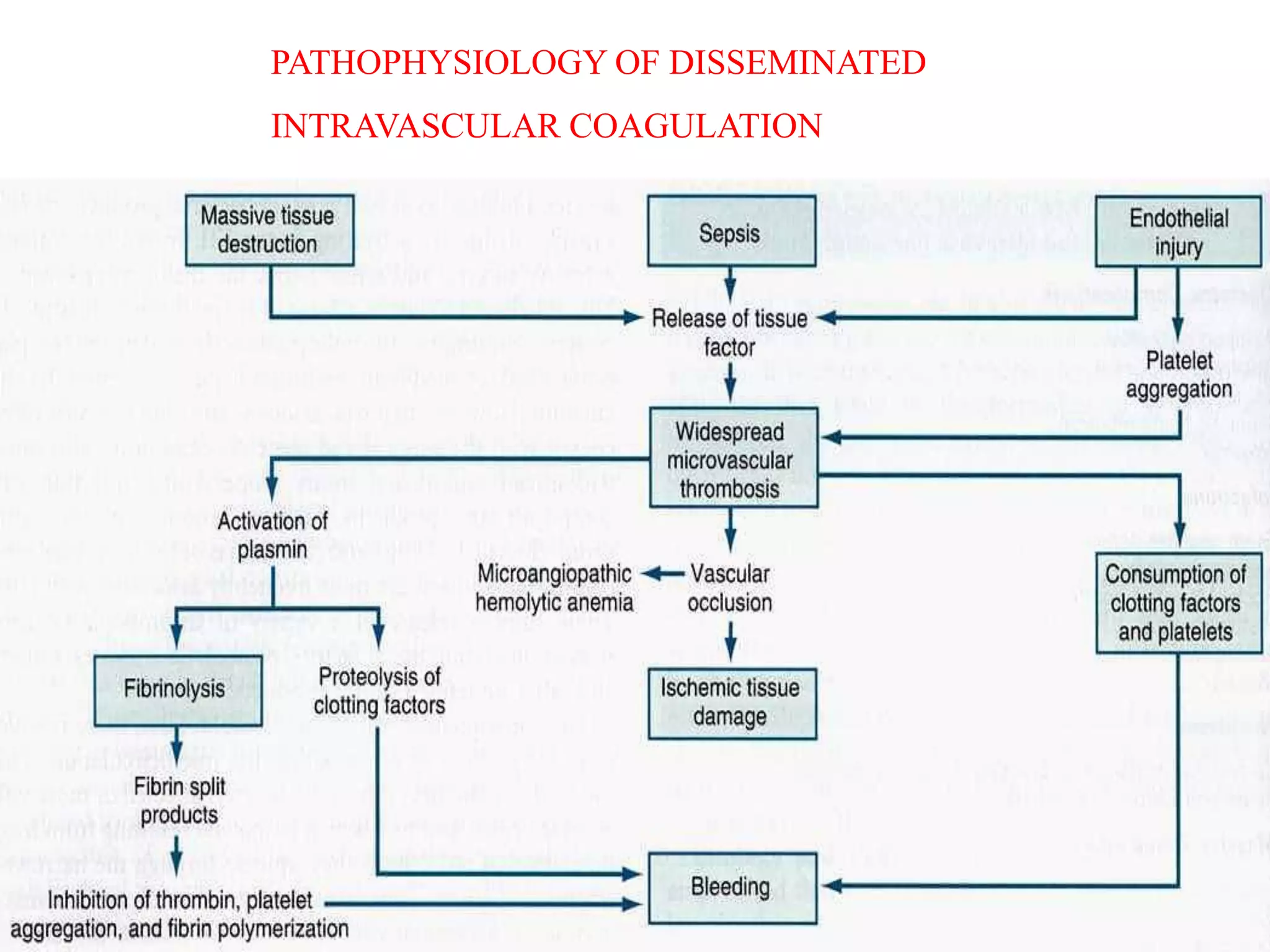
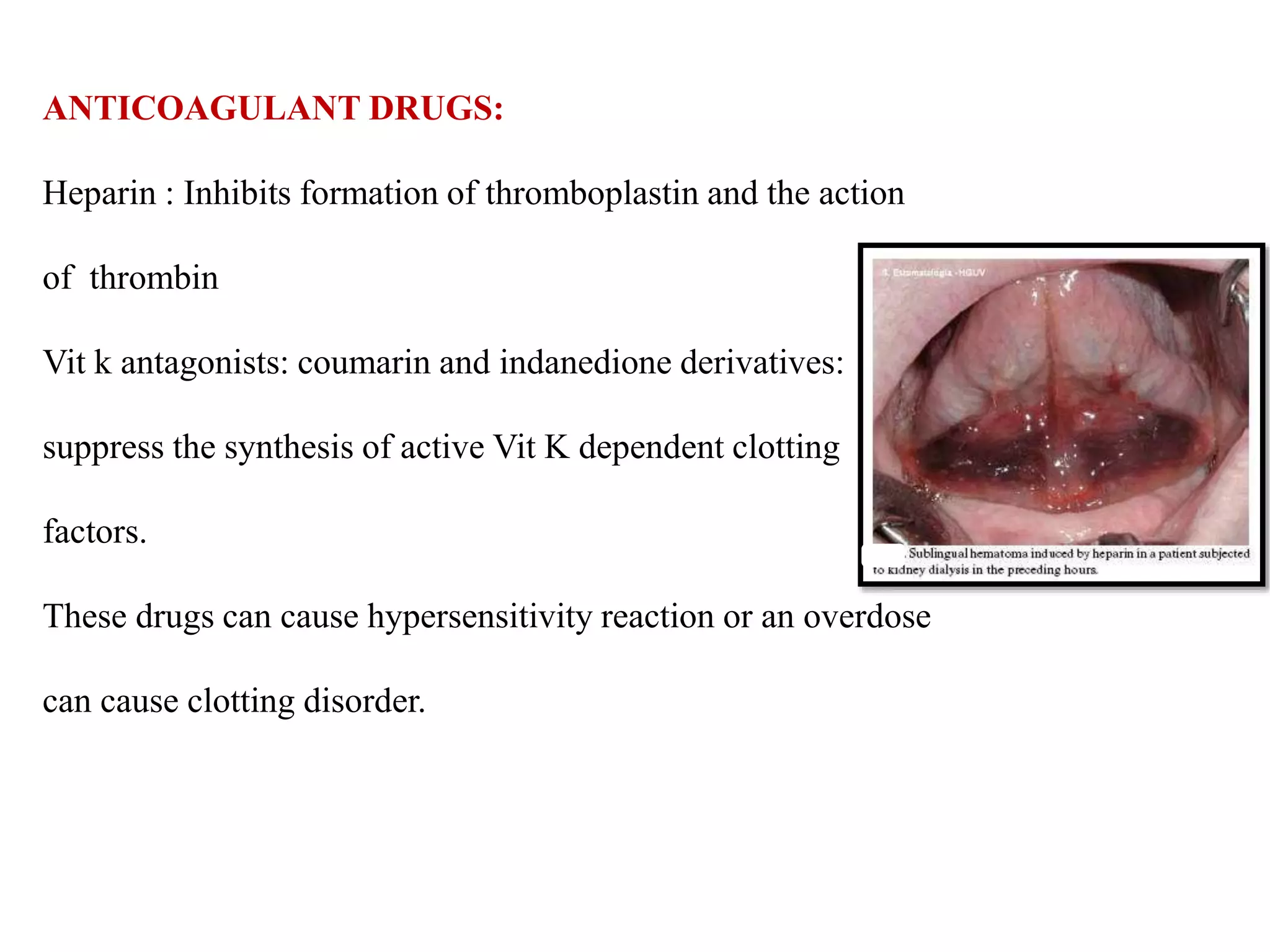
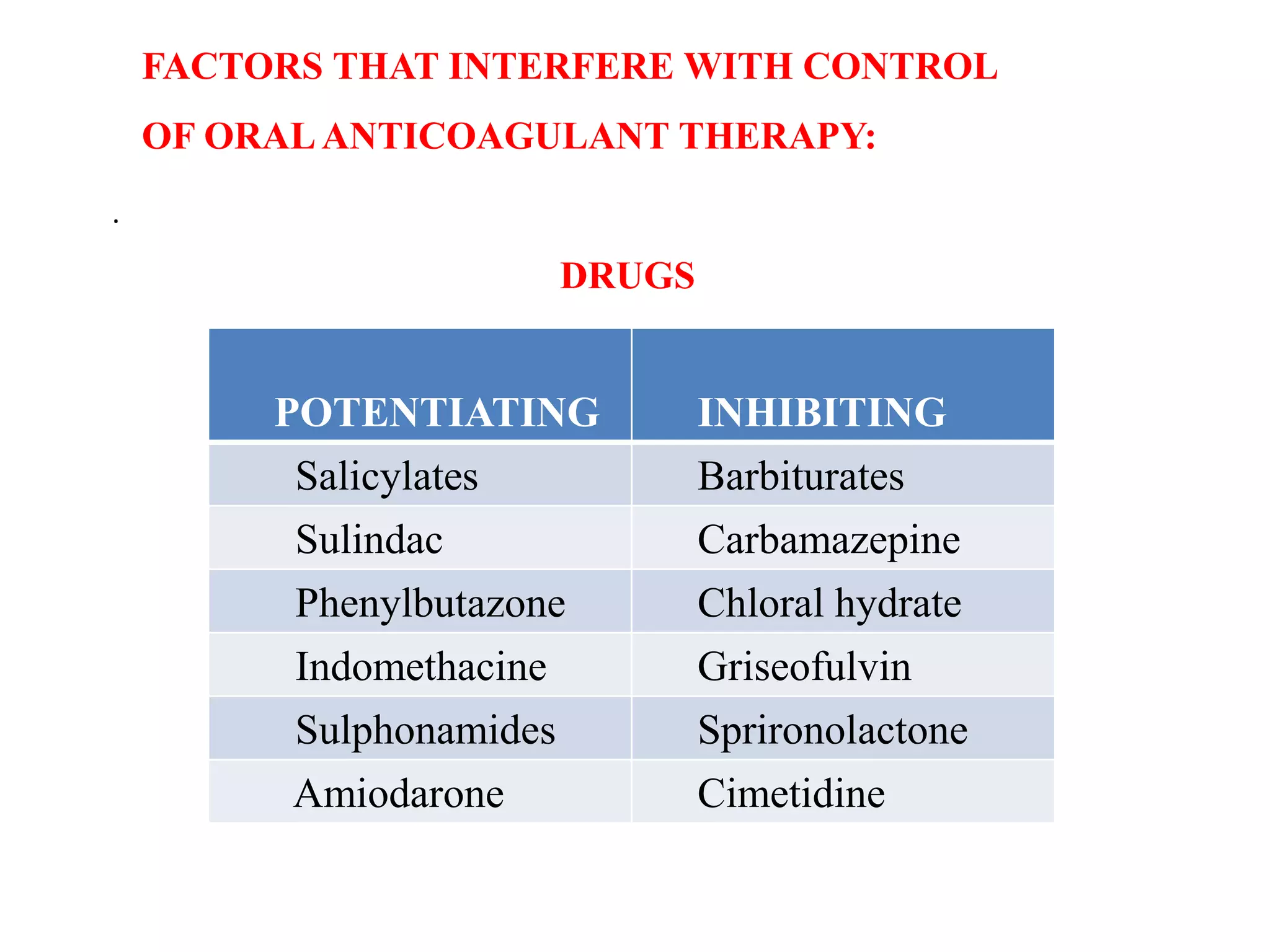
Diseases involving blood clotting factors can be inherited or acquired. The coagulation cascade involves several clotting factors that work together as enzymes to form a blood clot. Deficiencies or defects in these clotting factors can result in bleeding disorders. Some of the major clotting factor deficiencies include hemophilia A caused by factor VIII deficiency, hemophilia B caused by factor IX deficiency, von Willebrand disease caused by von Willebrand factor deficiency, and fibrinogen deficiencies that cause bleeding issues. Diagnostic tests evaluate clotting factor levels and clotting times. Treatment focuses on replacing the deficient clotting factor through blood products or concentrates.