
Downloaded 73 times



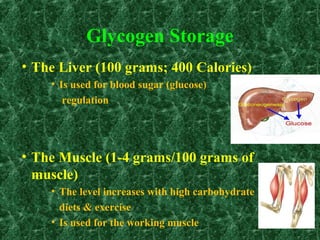

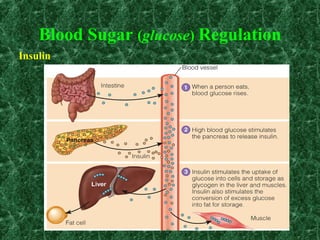

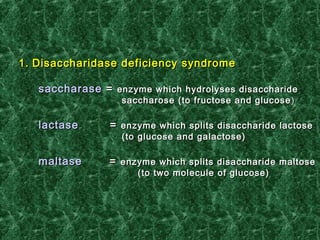


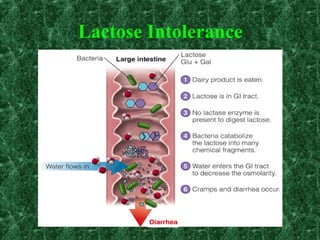

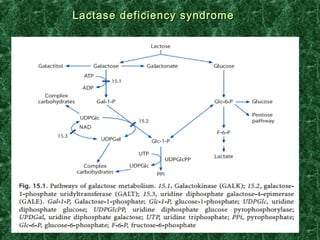




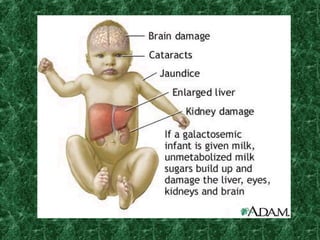












1) Glycogen storage diseases are inherited disorders caused by defects in glycogen metabolism enzymes, resulting in abnormal glycogen storage in tissues like the liver and muscle. 2) Symptoms vary depending on the type of enzyme defect and affected tissues, and can include hypoglycemia, hepatomegaly, muscle weakness, fatigue, and developmental delays. 3) The most common types are Von Gierke disease (type I) affecting glucose production in the liver, Pompe disease (type II) affecting heart and liver, and McArdle disease (type V) causing exercise intolerance due to a muscle enzyme defect.























































































