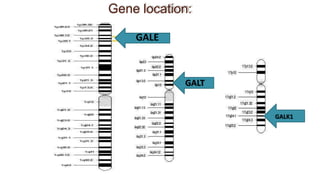
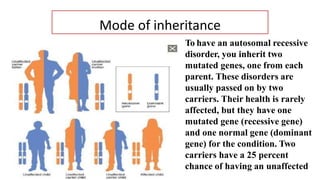
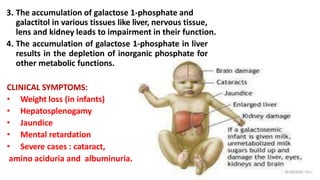
Galactosemia is a rare genetic metabolic disorder caused by a deficiency of the enzyme galactose-1-phosphate uridyltransferase, which is necessary for galactose metabolism. There are three main types depending on the specific enzyme deficiency. It is inherited in an autosomal recessive pattern and causes an inability to properly break down and use the sugar galactose. If left untreated, it can cause serious issues such as liver damage, cataracts, intellectual disabilities and more. Treatment involves a strict lifelong galactose-restricted diet.





































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)












