Downloaded 14 times

























![TypeXI, Fanconi-Bickelsyndrome
• Affected enzyme: Glucose transporter GLUT2 [solute
carrier family 2 ,facilitated glucose transporter]
• Clinical features: Similar features to Von Gierke's
disease, e.g. hypoglycaemia.](https://image.slidesharecdn.com/amerglycogenstoragedisease-201220161153/85/Amer-glycogen-storage-disease-27-320.jpg)

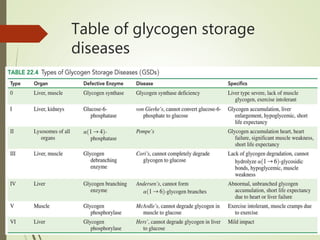







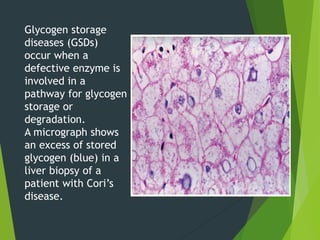
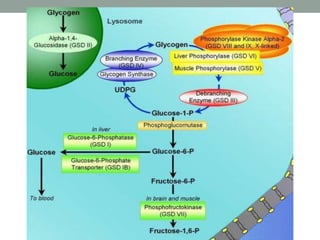
Glycogen storage disease (GSD) occurs due to defects in enzymes involved in glycogen synthesis or breakdown, leading to excess glycogen storage. There are 11 known types classified by the affected enzyme and tissue. Symptoms vary by type but can include hypoglycemia, liver and muscle involvement, and exercise intolerance. Treatment depends on type and may include dietary changes, enzyme replacement therapy, or liver transplantation. Prenatal testing is available for some types.






















































































