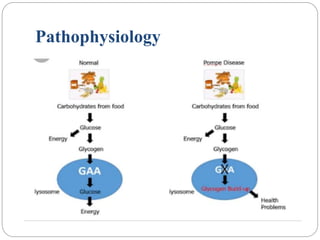
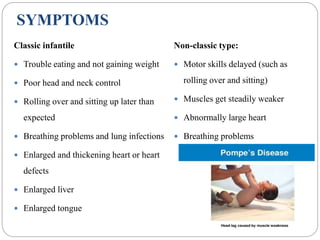
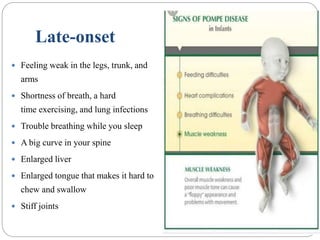
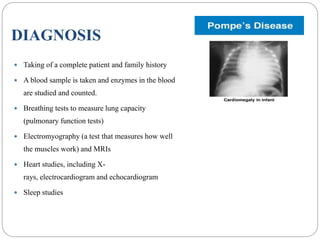
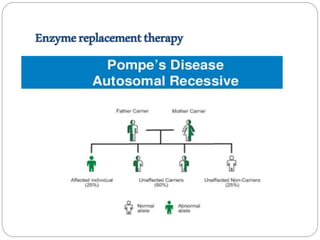
Pompe disease has three types: classic infantile-onset, non-classic infantile-onset, and late-onset, with varying appearance times and symptoms in infants, children, or adults. It is caused by a mutation in the gaa gene leading to acid maltase deficiency, resulting in sugar accumulation in muscles and organs, and can be inherited in an autosomal recessive pattern. Diagnosis involves enzyme tests and imaging, while treatment primarily includes enzyme replacement therapy alongside supportive care such as physiotherapy and dietary adjustments.