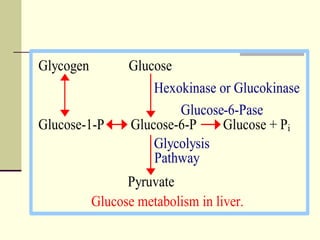
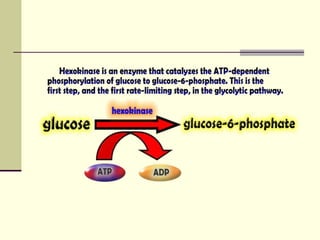
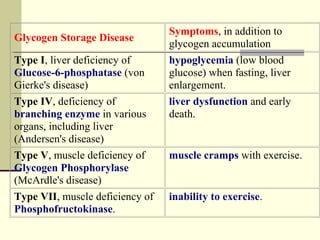
Glycogen storage diseases (GSDs) are genetic metabolic disorders caused by enzyme deficiencies affecting glycogen metabolism, leading to excess glycogen accumulation in cells. Symptoms can vary by GSD type and may include muscle cramps, liver enlargement, and hypoglycemia due to impaired glucose mobilization. The diagnosis is confirmed through enzyme activity tests, and treatment typically involves frequent meals to maintain normal blood glucose levels.