Download to read offline











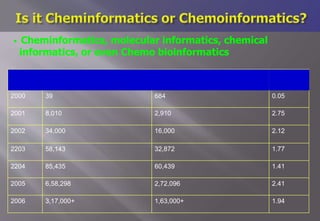



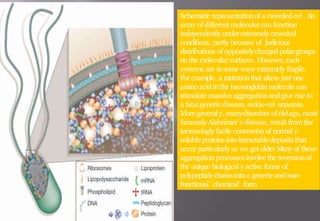








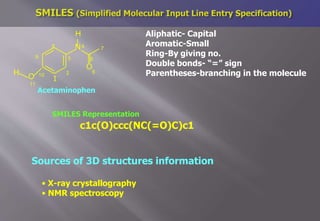





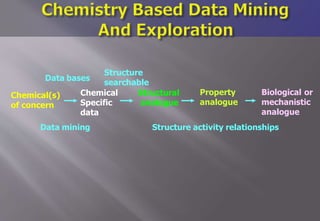





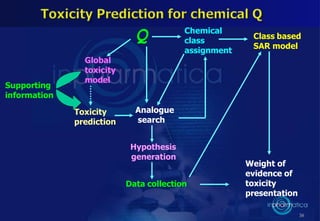


This document discusses cheminformatics and its applications. Cheminformatics combines chemistry and computer science to store and analyze chemical data for applications like drug discovery. It encompasses designing, organizing, analyzing and visualizing chemical information. Key topics covered include molecular representations, chemical databases, similarity searching, machine learning methods, and tools for molecular docking and drug discovery.























































































