Downloaded 55 times












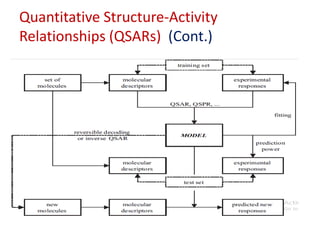
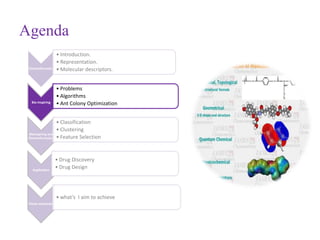


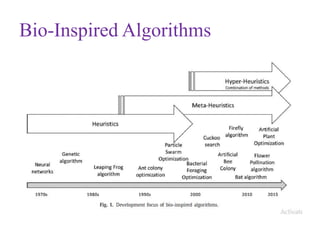

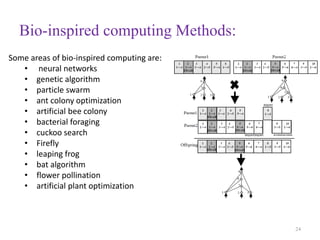






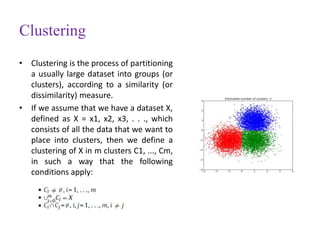



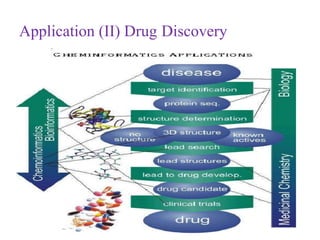
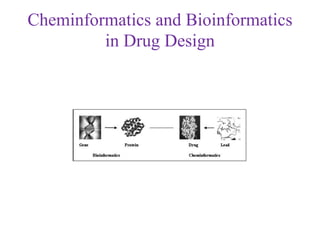







This document discusses applying bio-inspired computing techniques to problems in cheminformatics. It begins with introductions to cheminformatics and bio-inspired computing. Popular bio-inspired algorithms like ant colony optimization are explained. The document outlines applications of bio-inspired approaches to tasks in cheminformatics like classification, clustering, and feature selection. It concludes by noting potential applications in drug discovery and design.


















































































![5G Explained! A High Level Overview [Introduction]](https://cdn.slidesharecdn.com/ss_thumbnails/5gexplainedahighleveloverview-260119165306-cc137a3e-thumbnail.jpg?width=640&height=640&fit=bounds)




