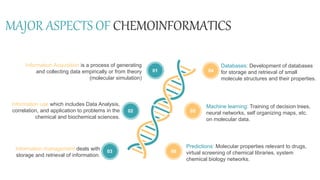
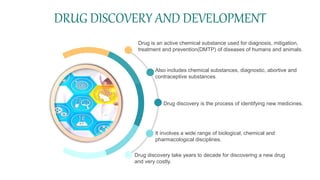
Cheminformatics combines chemistry, computer science, and information science to study large amounts of chemical information, mostly with computer assistance. It encompasses the design, creation, organization, storage, retrieval, analysis, and use of chemical data. Cheminformatics has various applications including drug discovery. It uses tools like databases, machine learning, molecular properties predictions, and information analysis to help identify new drug leads. Future trends include increased data integration, computer-assisted synthesis design, and expanded use of cheminformatics methods in theoretical chemistry and protein studies. Cheminformatics plays an important role in modern drug development.