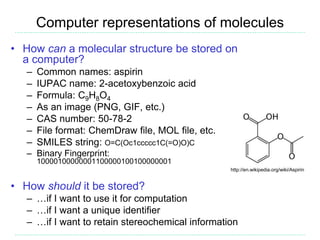
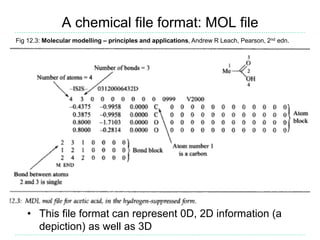
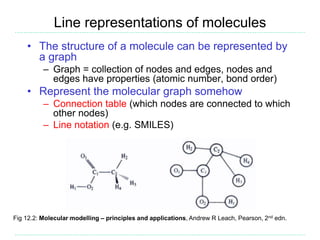
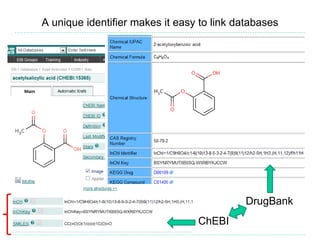
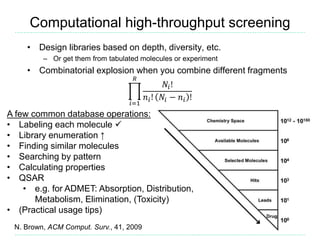
The document provides an overview of cheminformatics, focusing on the representation, management, and use of chemical information in computational drug discovery. It discusses various molecular representation formats, such as SMILES and InChI, along with techniques for compound selection, molecular similarity, and quantitative structure-activity relationships (QSAR). Key takeaways highlight the importance of unique identifiers for chemical data and the utility of cheminformatics in enhancing drug discovery processes.





(I)F means that looking from the Br, the Cl, I, and
F are arranged anticlockwise
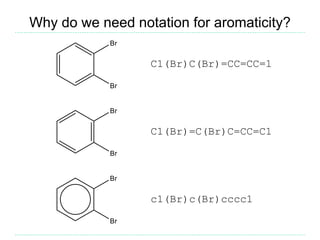
• To represent aromaticity, use lower case
• C1CCCCC1 (cyclohexane)
• c1ccccc1 (benzene)
Cl
C C
Br](https://image.slidesharecdn.com/cheminformatics20180306-180309150509/85/Overview-of-cheminformatics-10-320.jpg)





[#6]
• Any oxygen with exactly two bonds each to a carbon
• Can get (a lot) more complicated
– Carbonic Acid or Carbonic Acid-Ester:
[CX3](=[OX1])([OX2])[OX2H,OX1H0-1]
• Hits acid and conjugate base. Won't hit carbonic acid diester
– Many good examples online:
http://www.daylight.com/dayhtml_tutorials/languages/smarts/smarts_examples.html
• Examples of use
– Filtering structures
– Identify substructures that are associated with toxicological
problems
– Develop or use a group contribution descriptor such as TPSA](https://image.slidesharecdn.com/cheminformatics20180306-180309150509/85/Overview-of-cheminformatics-21-320.jpg)
([OX2])[OX2H,OX1H0-1]](https://image.slidesharecdn.com/cheminformatics20180306-180309150509/85/Overview-of-cheminformatics-22-320.jpg)



![Open Source cheminformatics software resources
• GUI:
– Open Babel, Avogadro
– LICSS – Excel-CDK interface
• Command-line interface:
– Open Babel (“babel/obabel”)
– MayaChemTools
• Programming toolkits:
– Open Babel (C++, Perl, Python, .NET, Java), RDKit (C++, Python),
Chemistry Development Kit [CDK] (Java, Jython, ...), PerlMol (Perl),
MayaChemTools (Perl)
– Cinfony (by Noel!) presents a simplified interface to some of these
– Javascript libraries
– Materials simulation libraries: Pymatgen, ASE (Python)
• Specialized toolkits:
– OSRA: image to structure
– OPSIN: name to structure
– OSCAR: Identify chemical terms in text](https://image.slidesharecdn.com/cheminformatics20180306-180309150509/85/Overview-of-cheminformatics-28-320.jpg)
![Getting started with Open Babel
• Convert formats
– # -O specifies a file, -o specifies a format and prints to stdout by default
– obabel raspa_framework.pdb -O framework.cif
• Many import formats have input (-a) or output (-x) flags
– Open Babel detects bonds by default (sometimes expensive). Can
sometimes disable using the flag -ab
– https://openbabel.org/docs/dev/FileFormats/Overview.html
• Draw an SVG (or PNG) line structure, highlighting a pattern
– obabel caffeine.smi -O caffeine.svg -xe -xC -s "[#7][CH3]" "purple"
• Convert SMILES to InChIKey
– obabel -:"CC(=O)Cl" -oinchikey
• Get molecular properties
– obprop my_file.smi
• Make sure BABEL_DATADIR variable is set properly
• See also the Python module: import pybel, openbabel
– Warning: install the package openbabel to get both of these, NOT pybel
– Interoperable with rdkit, another powerful cheminformatics library](https://image.slidesharecdn.com/cheminformatics20180306-180309150509/85/Overview-of-cheminformatics-29-320.jpg)