Downloaded 280 times
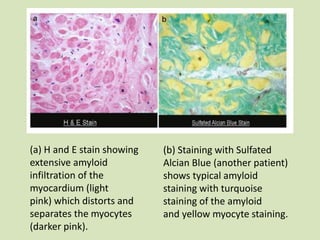

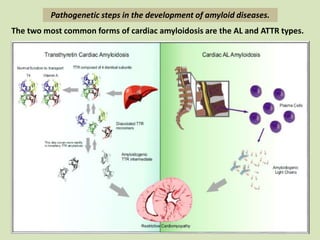
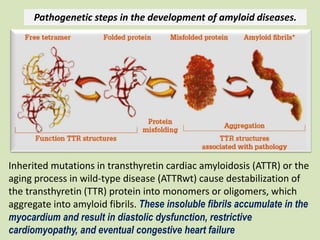


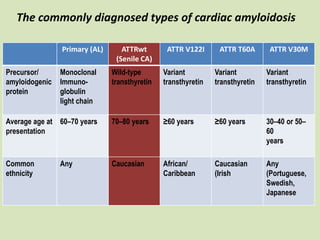
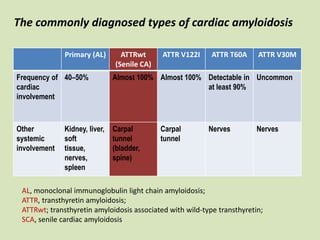







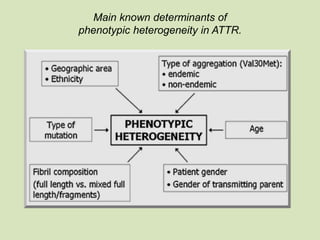








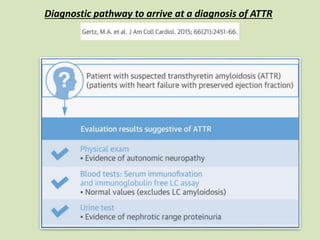

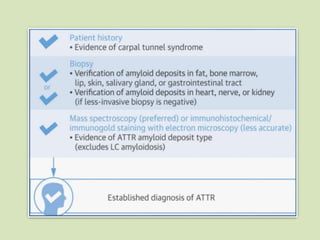
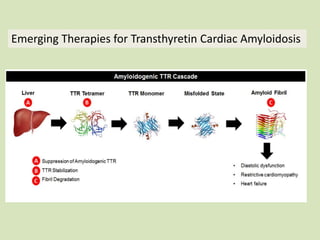
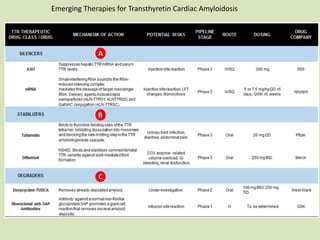



This document discusses cardiac amyloidosis, a group of disorders characterized by the aggregation of misfolded proteins into amyloid fibrils, which can lead to heart dysfunction. Two common types are AL and ATTR amyloidosis, with discussions on their pathogenesis, clinical manifestations, and challenges in diagnosis. The paper emphasizes the importance of high suspicion for amyloidosis in patients with heart failure and other symptoms, highlighting the need for accurate diagnostic pathways to enhance diagnosis and treatment.
























































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)


















