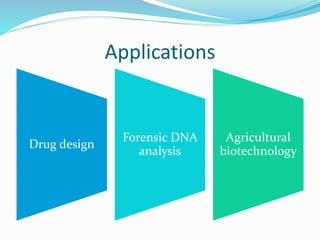
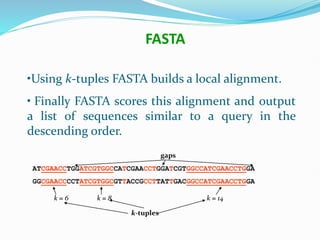
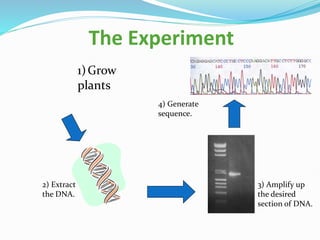
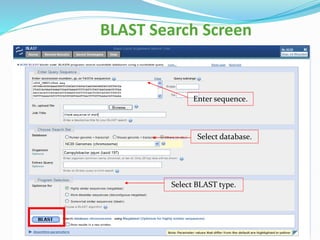
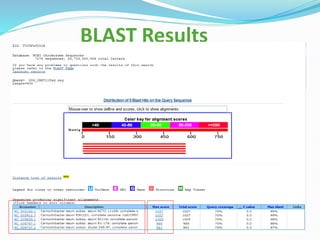
Bioinformatics is a multidisciplinary field that applies computational techniques to manage and analyze biological data, focusing on molecular sequences, structures, and functions. It plays a crucial role in areas such as drug design, DNA analysis, and agricultural biotechnology, utilizing tools like FASTA and BLAST for sequence similarity searches. The document also outlines the historical context and practical applications of bioinformatics in research.