
Download to read offline











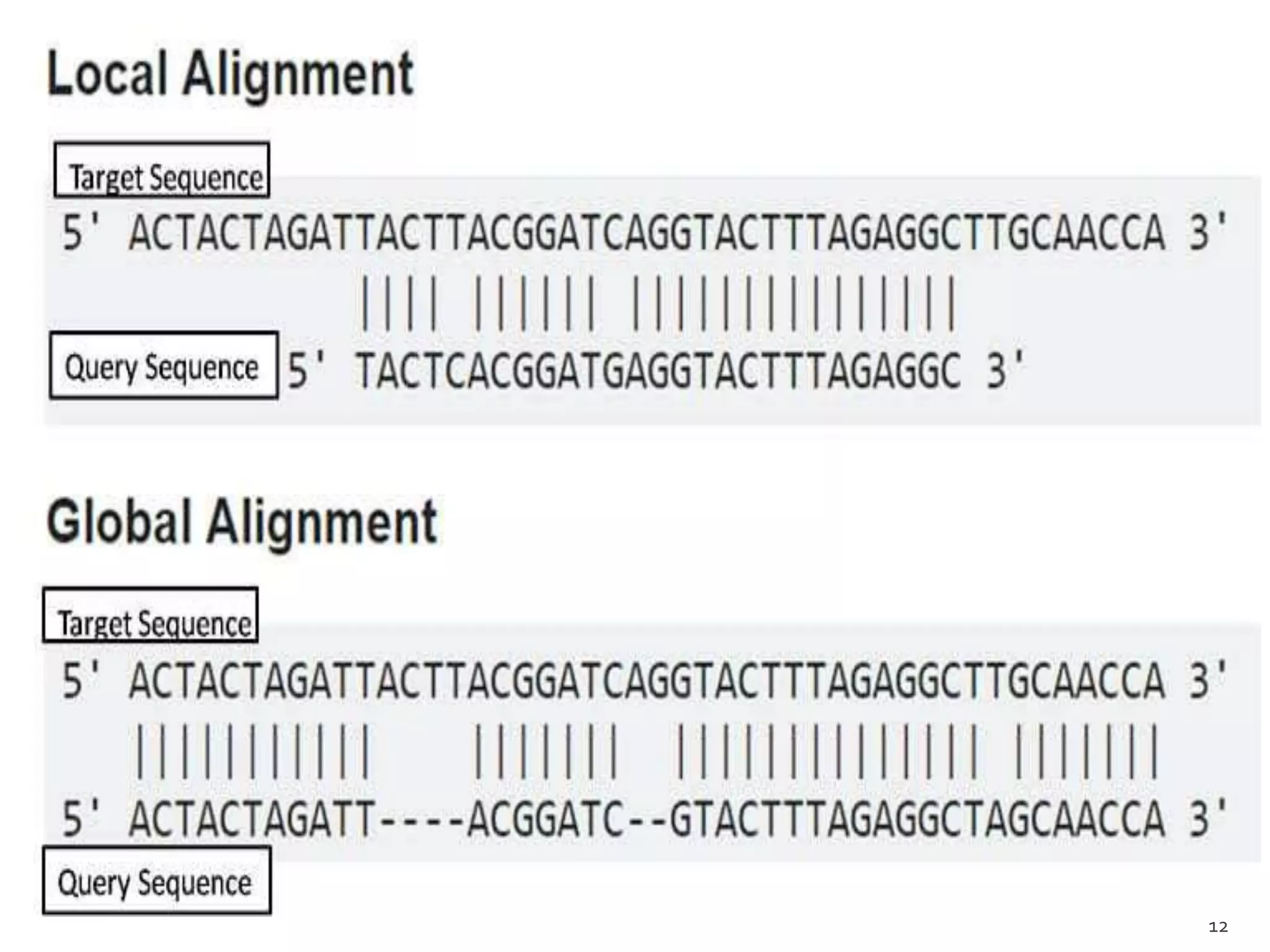







This document discusses sequence alignment, which involves arranging biological sequences like DNA, RNA, or proteins to identify regions of similarity. It covers the basic concepts of sequence alignment including global versus local alignment and different methods like dot matrix, dynamic programming, and word-based approaches. Dynamic programming is highlighted as the most common algorithm that uses a scoring system to find the optimal alignment between two sequences.




















































































