Downloaded 65 times















![ First: Calculate bit score
S = Score of the alignment (Raw Score)
, values depend on the scoring scheme and
sequence composition of a database.
[log value is natural logarithm (log base e)]
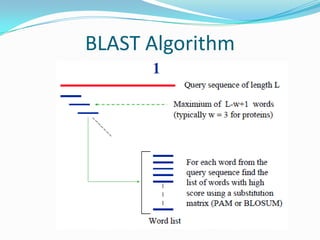
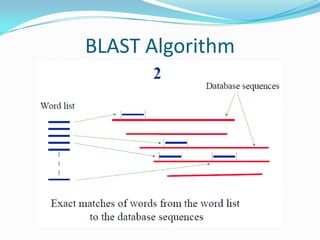
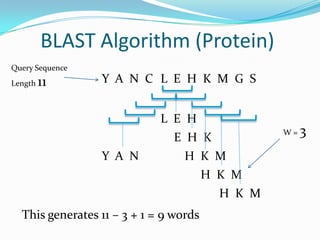
BLAST Algorithm
E-Value Calculation](https://image.slidesharecdn.com/blast20131-130630220412-phpapp01/85/Blast-2013-1-21-320.jpg)









![Homework 4A
Determine the common proteins in Domestic Cat
[Felis catus], Tiger [Panthera tigris] and Snow Leopard
[Uncia uncia] using this initiating sequence
>gi|145558804
MSMVYINMFLAFIMSLMGLLMYRSHLMSSLLCLEGMMLSLFIMMTVAILNNHFTLASMTPII
LLVFAACEAALGLSLLVMVSNTYGTDYVQNLNLLQC
Report for each protein match: Protein
name, accession number, bit score, raw score, E-
Value, Identities, Positives and Gaps.](https://image.slidesharecdn.com/blast20131-130630220412-phpapp01/85/Blast-2013-1-32-320.jpg)
![Homework 4B
H5N1 is the subtype of the Influenza A Virus which is a
bird-adapted strain. This subtype can cause “avian
influenza” or “bird flu” which is fatal to human.
Use DNA sequence with GenBank Accession number
JX120150.1 as a seed sequence to search for other TWO
matching sequences, each belonging to a different
Influenza A virus subtypes (HXNX). [Use Blastn]
Report for each subtype match: Subtype
name, Organism origin, Sequence name, accession
number, bit score, raw score, E-
Value, Identities, Positives and Gaps](https://image.slidesharecdn.com/blast20131-130630220412-phpapp01/85/Blast-2013-1-33-320.jpg)
![Homework 4C
Suppose you have acquired an unknown protein
sequence
FLWLWPYLSYIEAVPIRKVQDDTKTLIKTIVTRINDISHTQAVSSKQRVAGLDFIP
GLHPVLSLSRMDQTLAIYQQILTSLHSRNVVQISNDLENLRDLLHLLASSKS
(1) Use BLAST program to find out which species this sequence most likely
belongs to.
(2) Report both scientific and common name for the species.
(3) This sequence matches to a certain protein of that species, Report E-Value,
protein accession number [GenBank], Protein name, Length, Full sequence
and Function.](https://image.slidesharecdn.com/blast20131-130630220412-phpapp01/85/Blast-2013-1-34-320.jpg)



This document provides an overview of the BLAST algorithm used for comparing biological sequences and identifying sequence similarities. It describes how BLAST works by generating words from a query sequence and searching a database for exact matches. Significant matches are extended locally to identify Maximal Segment Pairs (MSPs) based on scoring. MSPs are evaluated and ranked using E-values, which estimate the statistical significance of matches. The document also discusses different BLAST programs and provides examples of running BLAST searches and interpreting results. Homework assignments are included applying BLAST to specific sequence analysis tasks.























































































