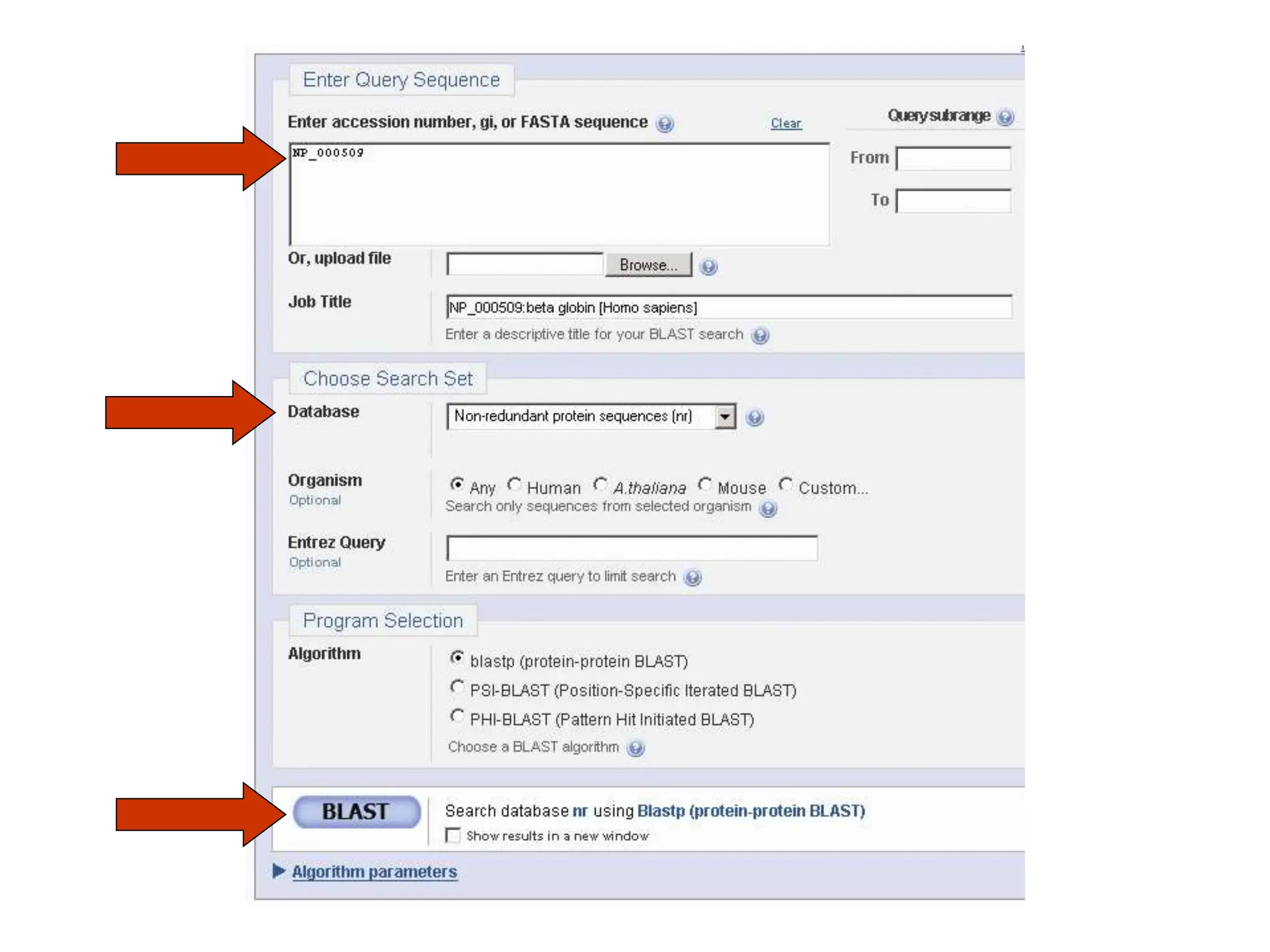
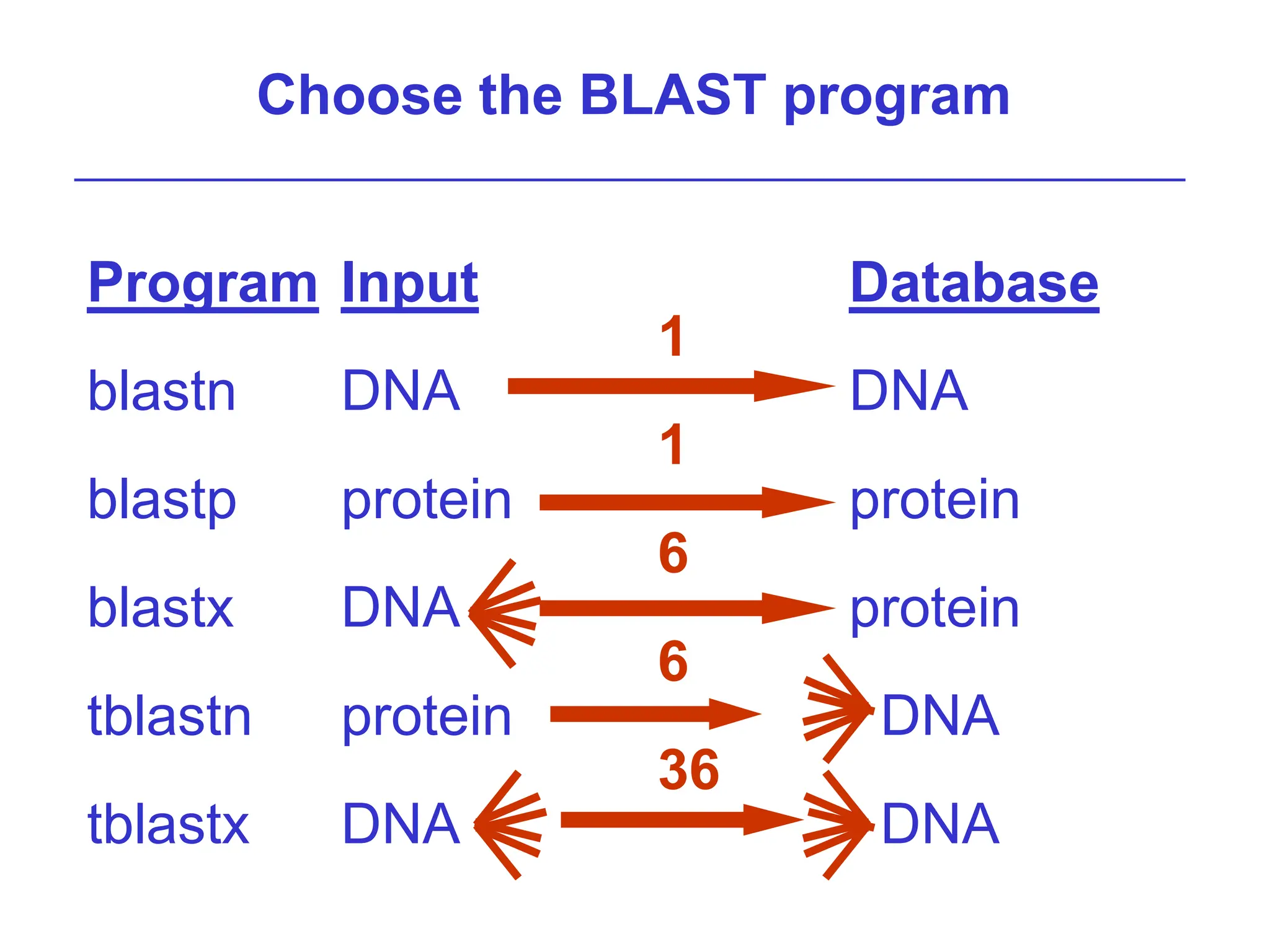
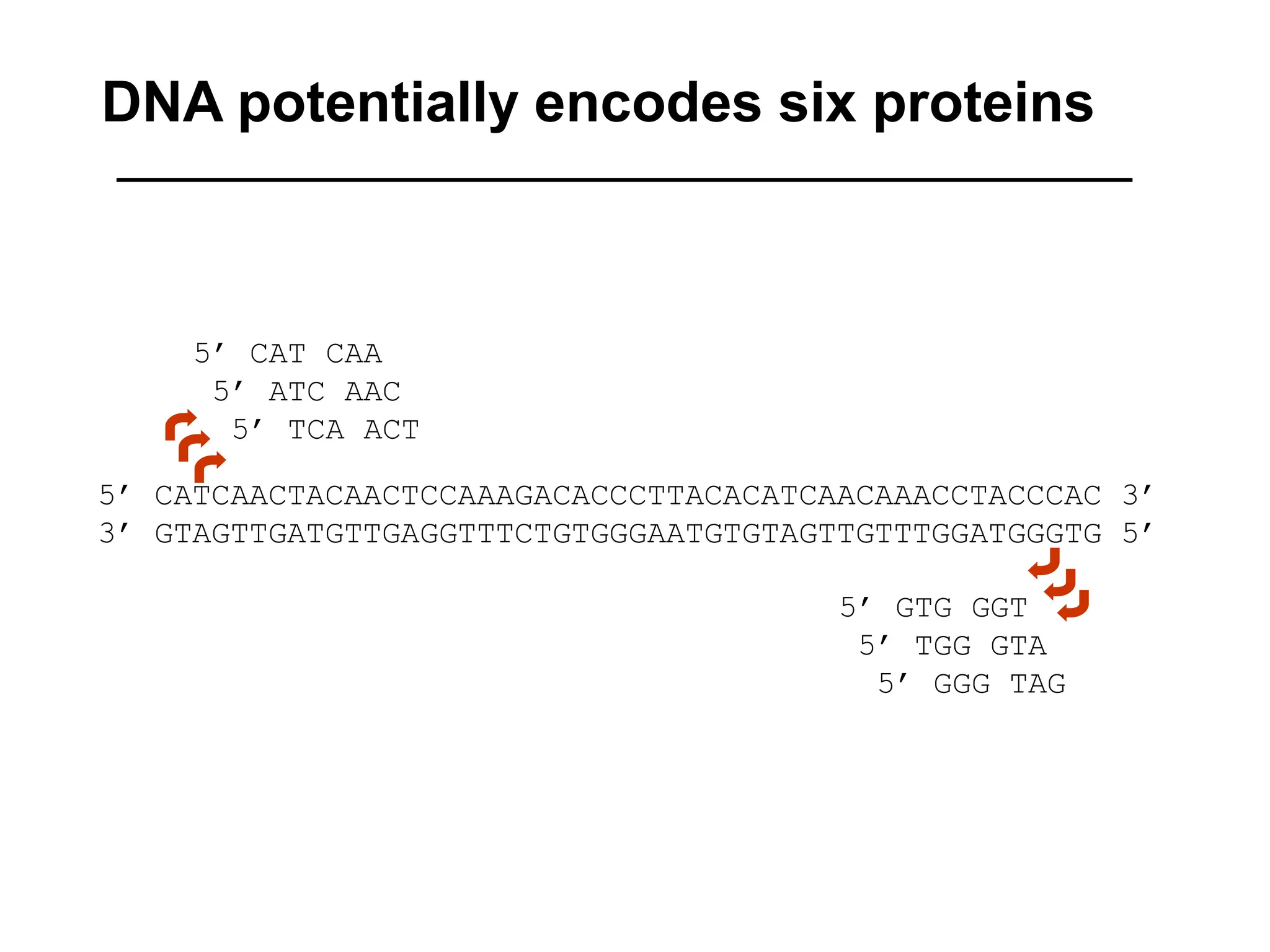
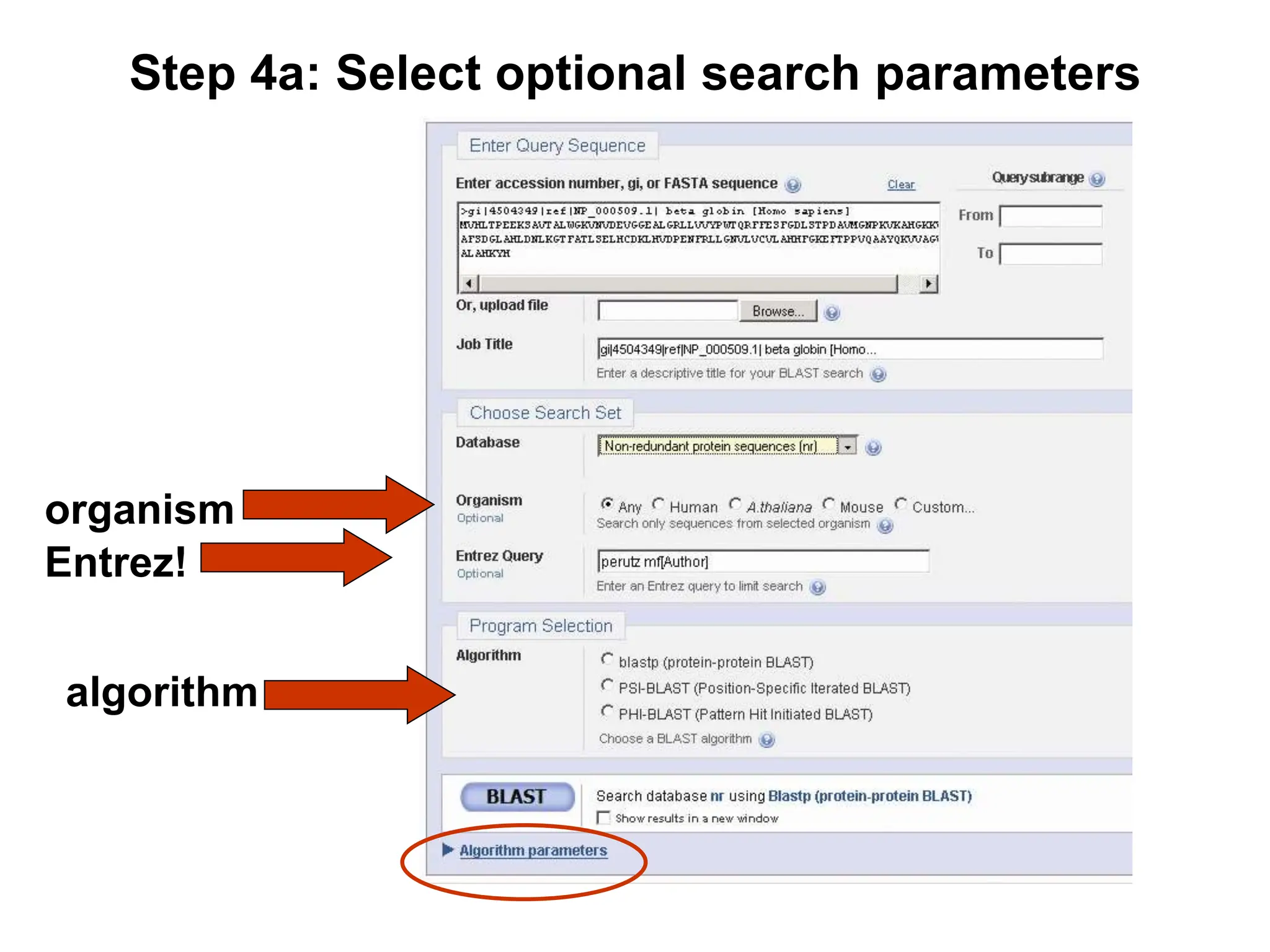
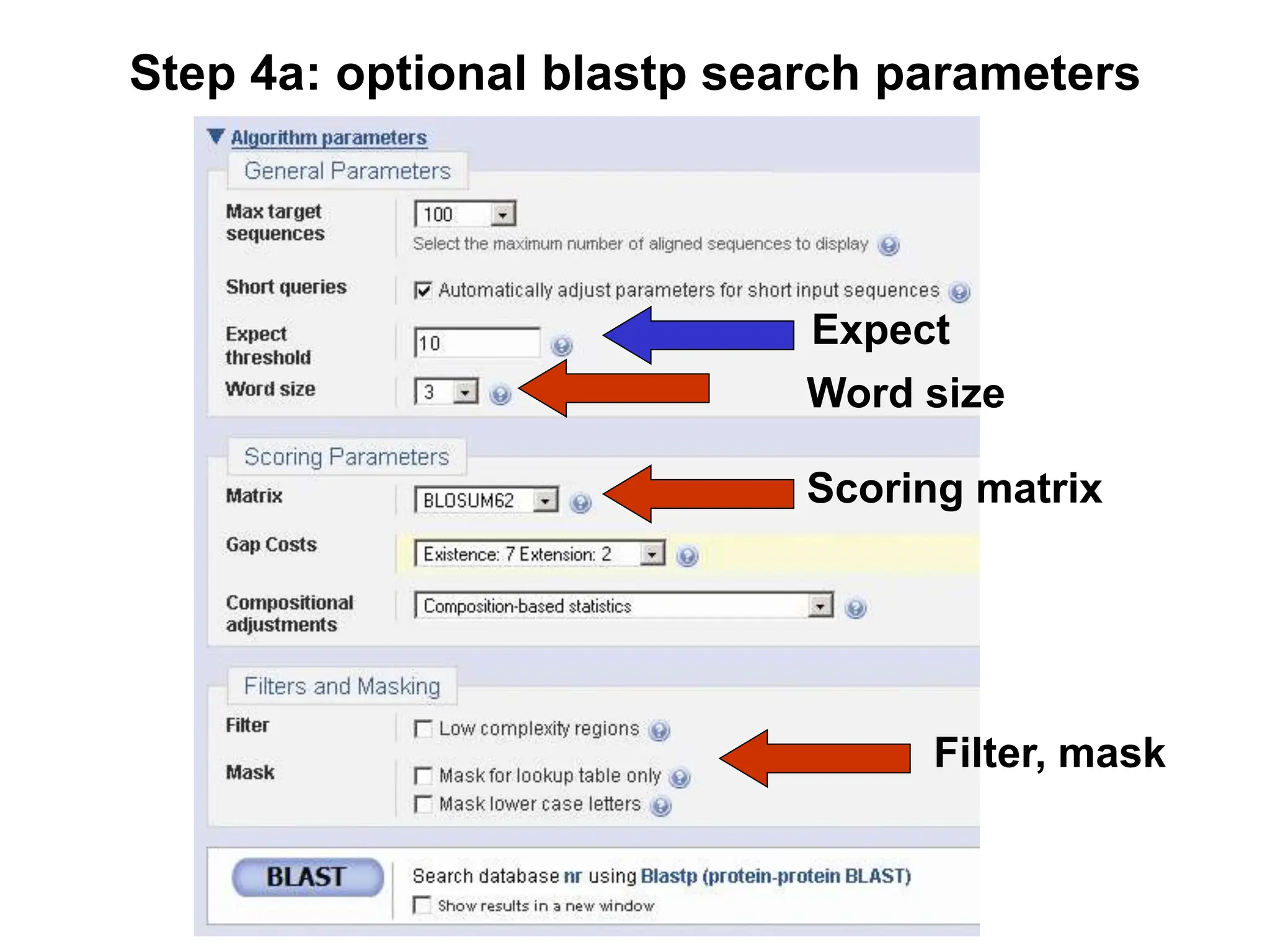
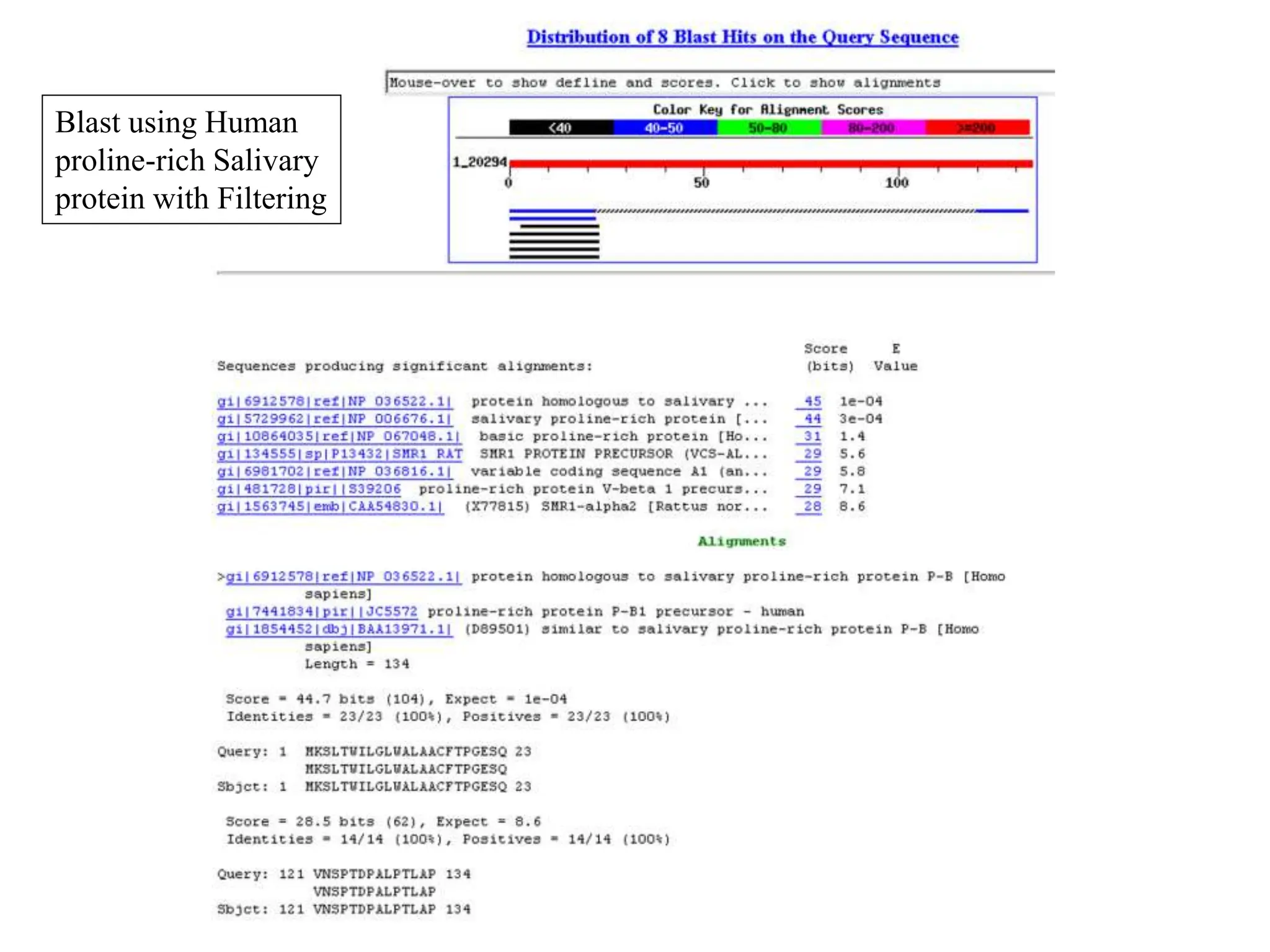
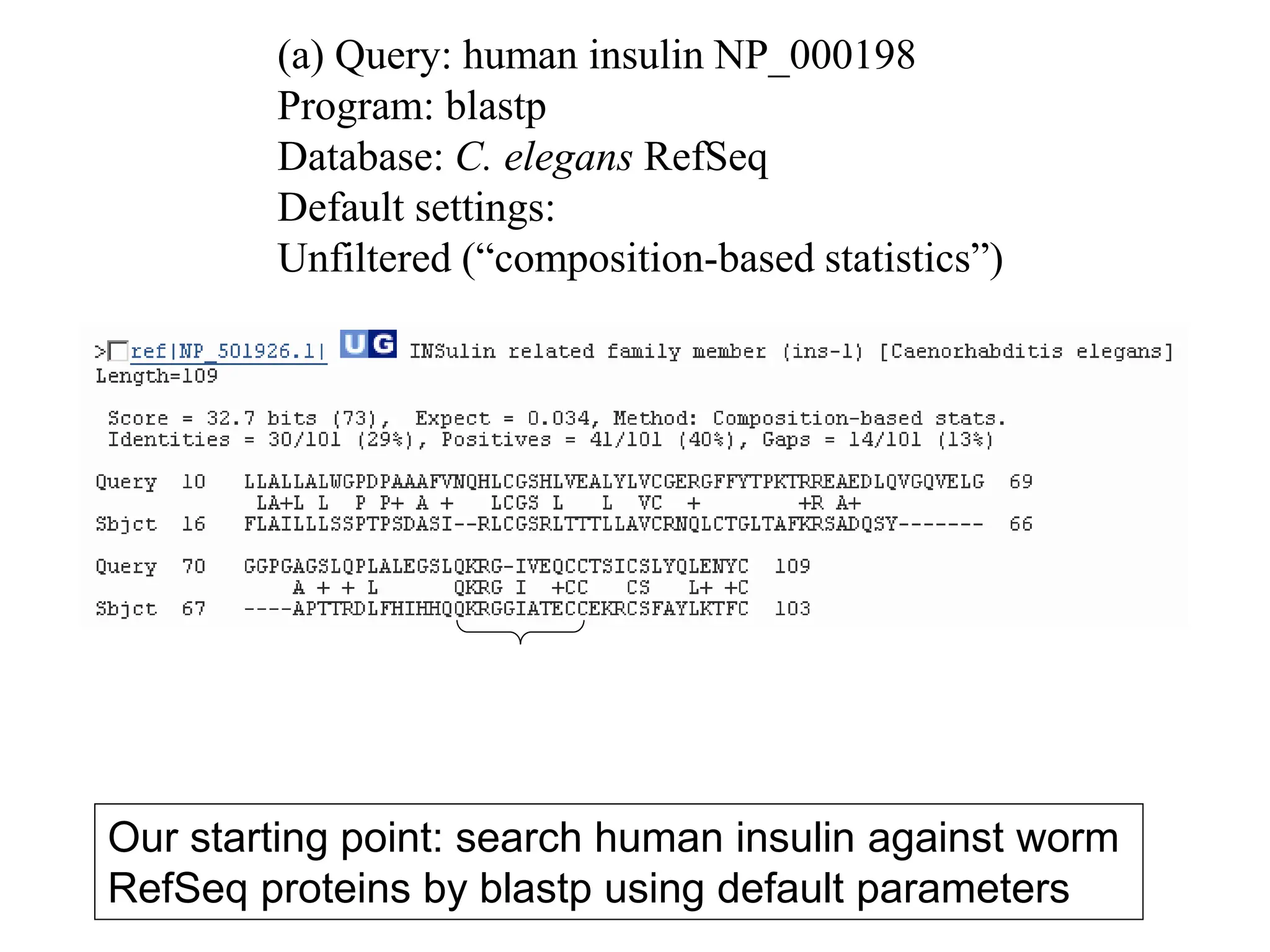
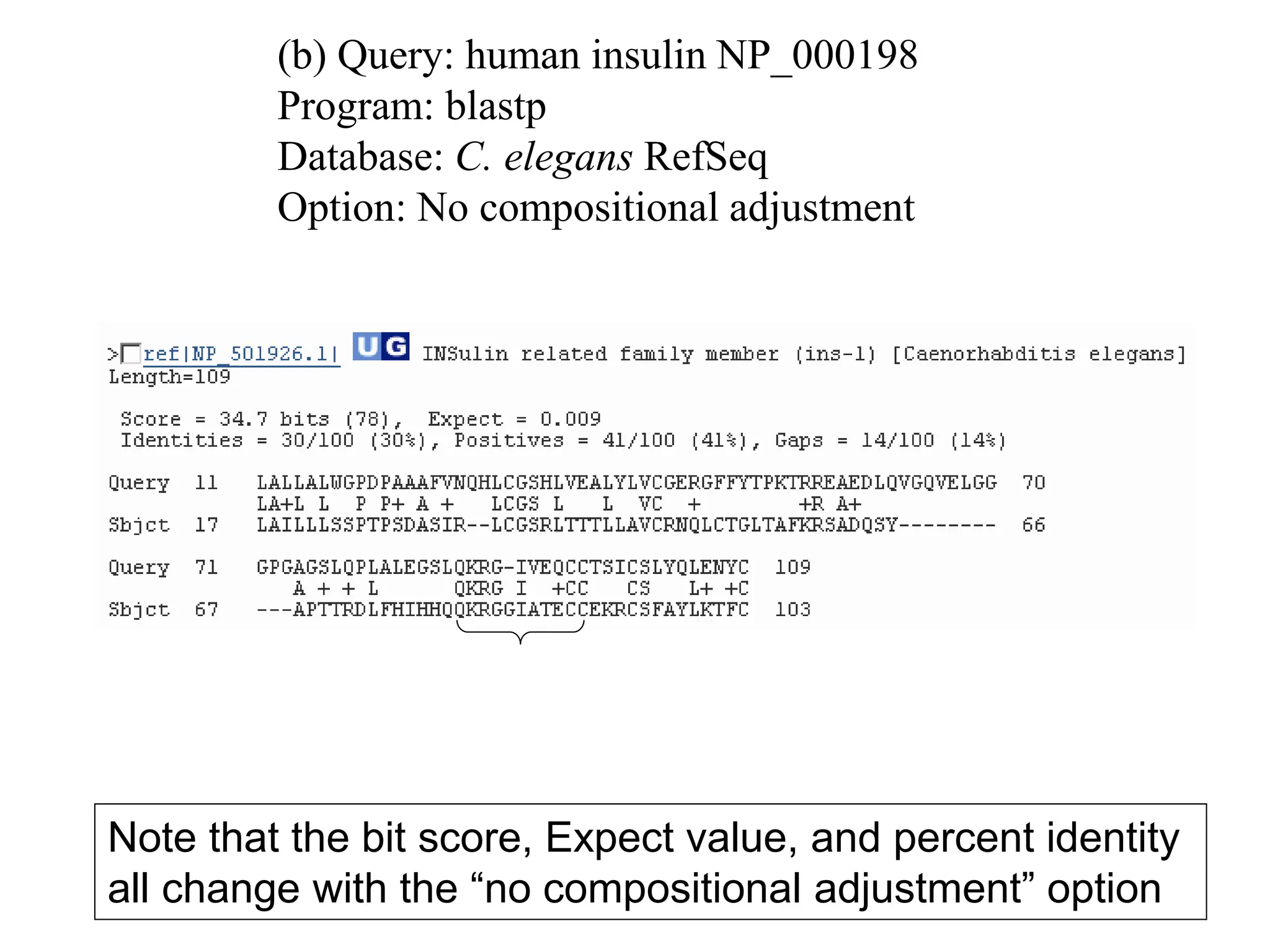
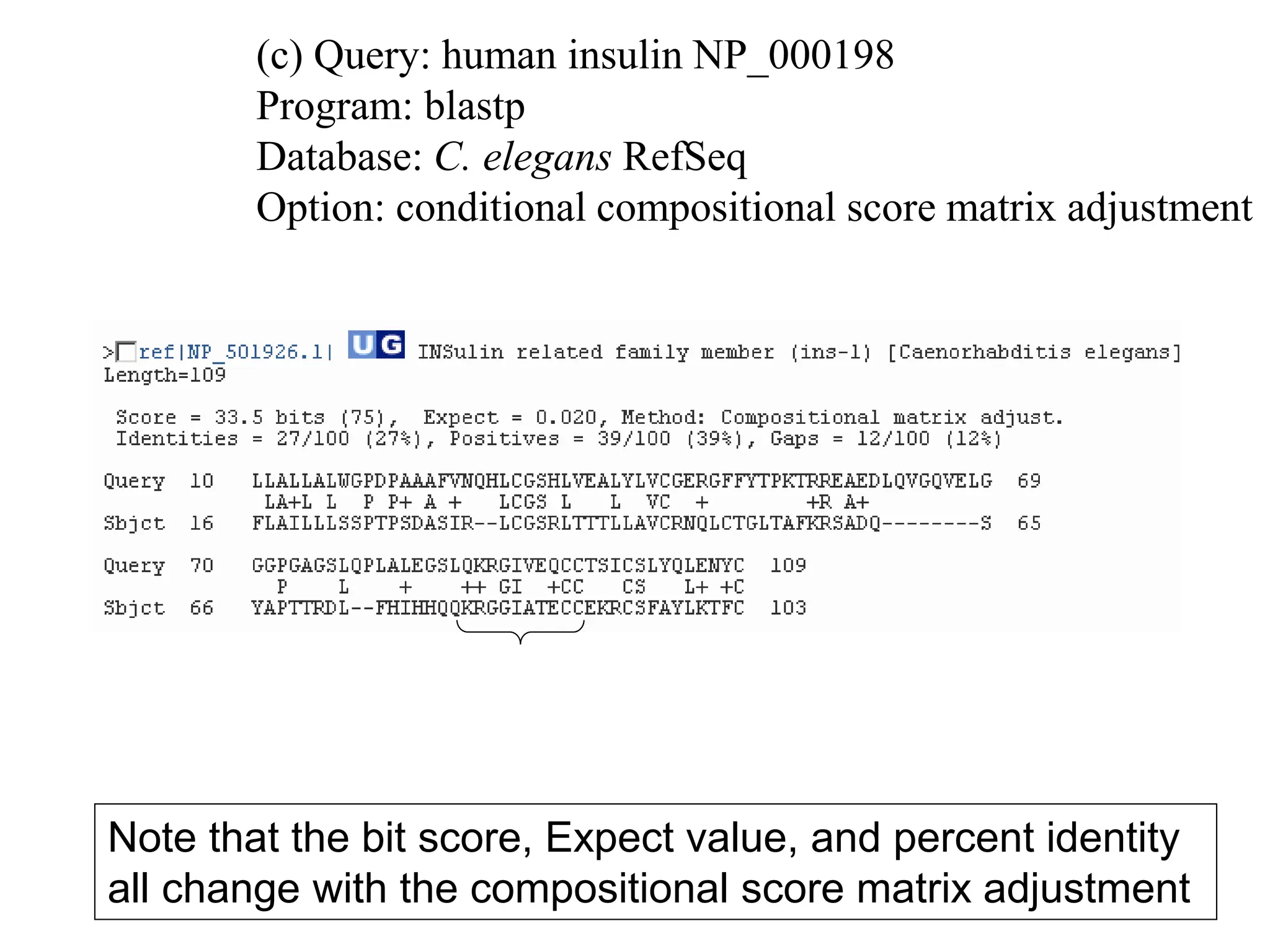
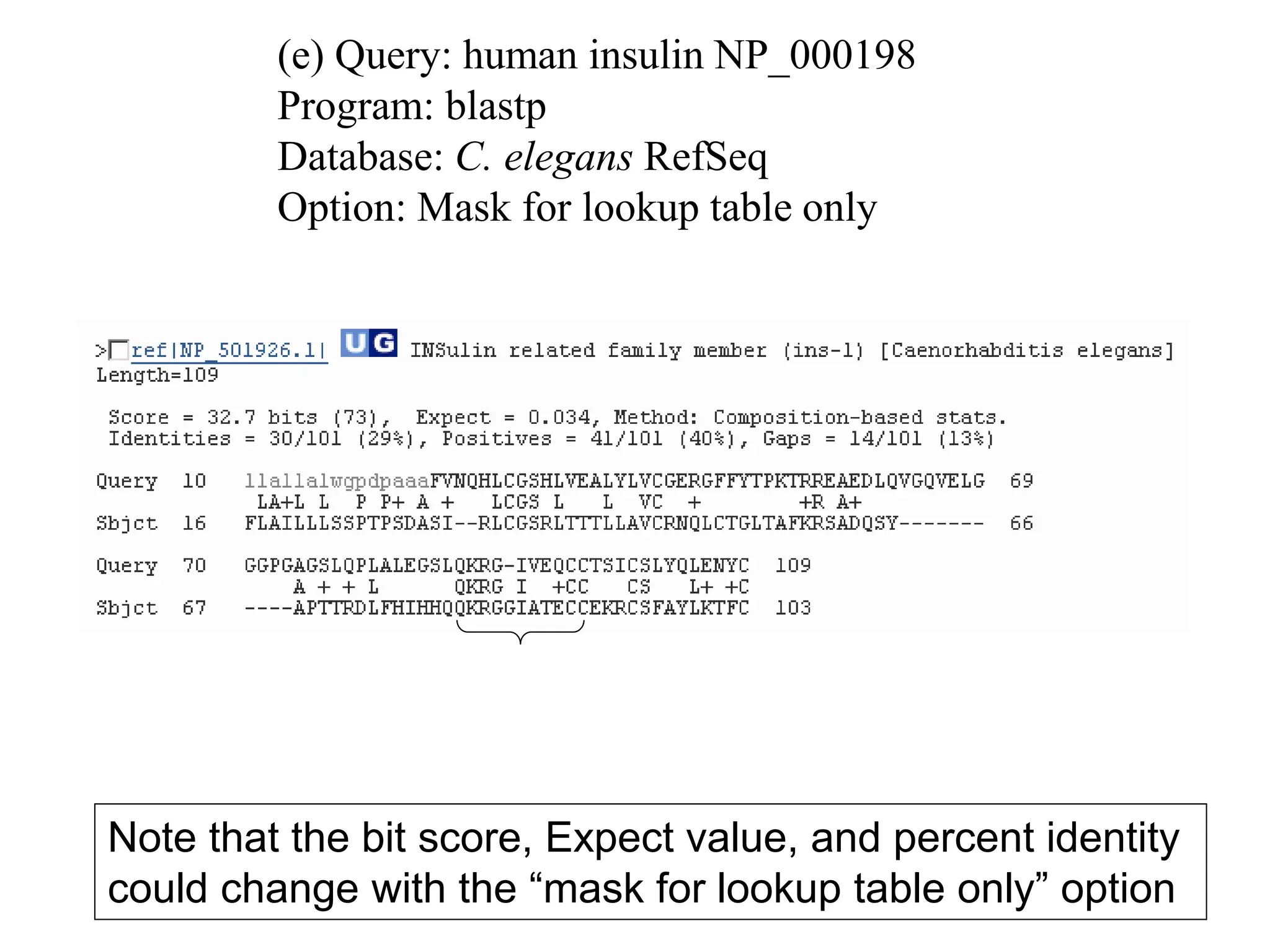
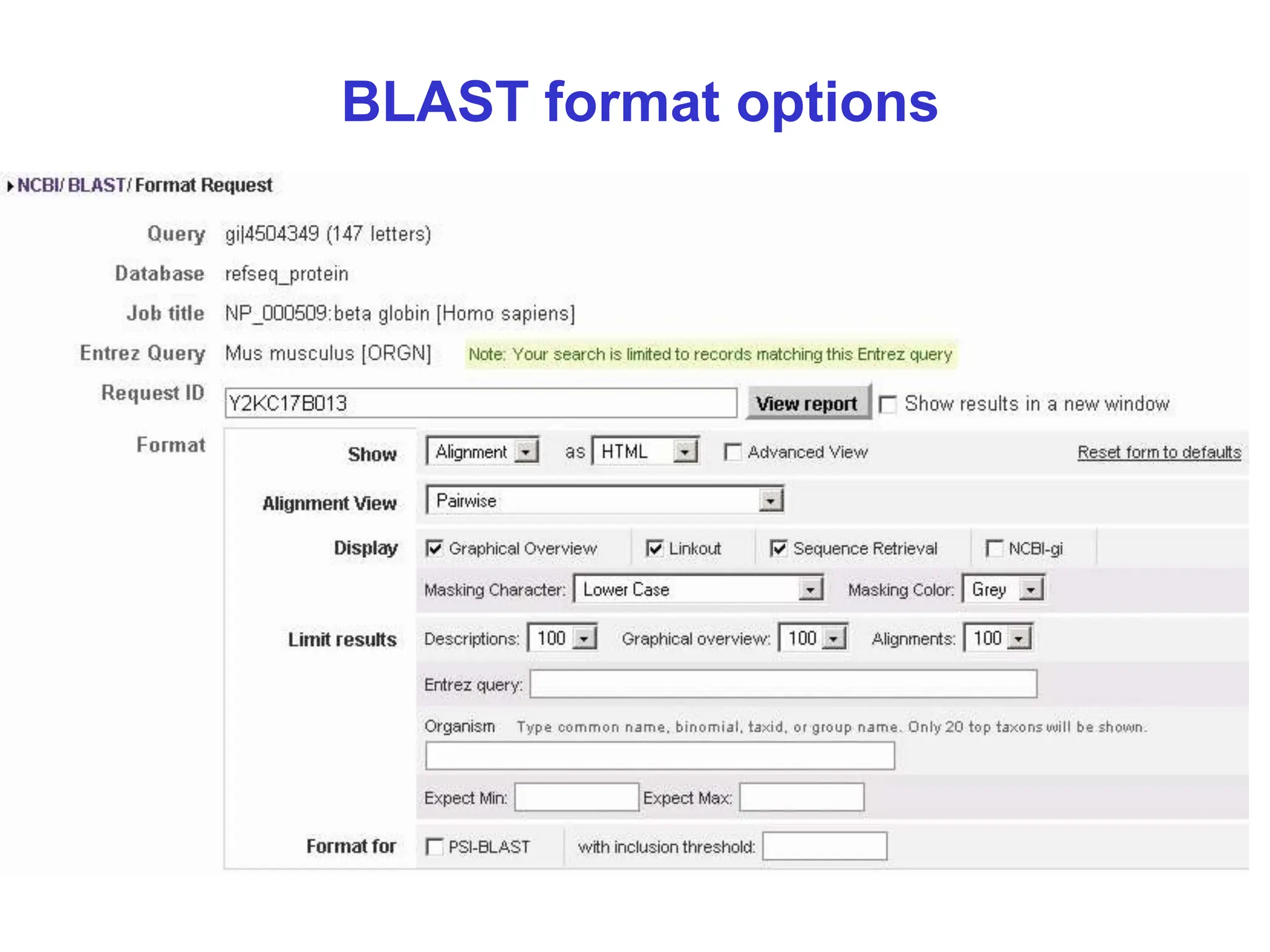
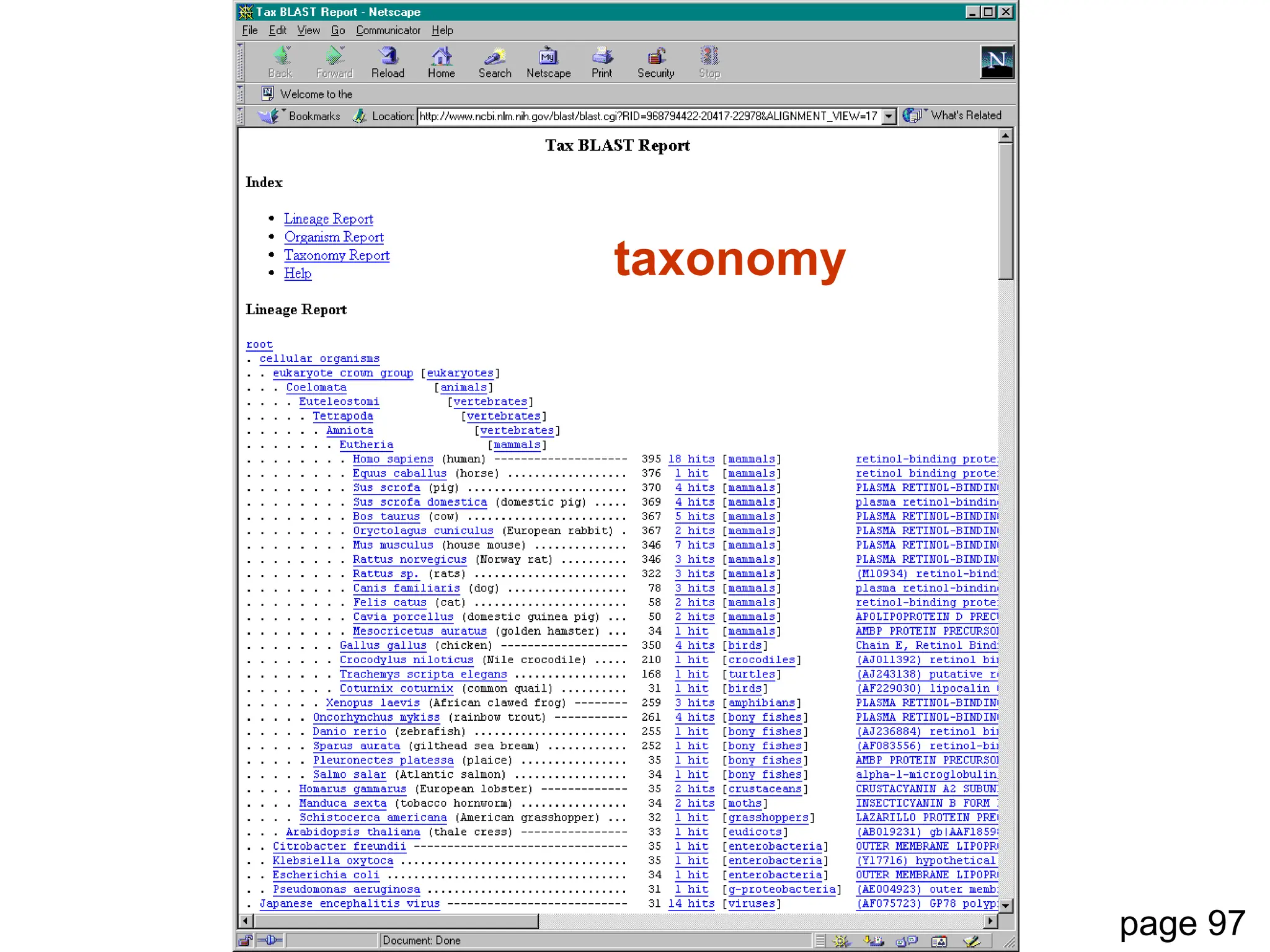
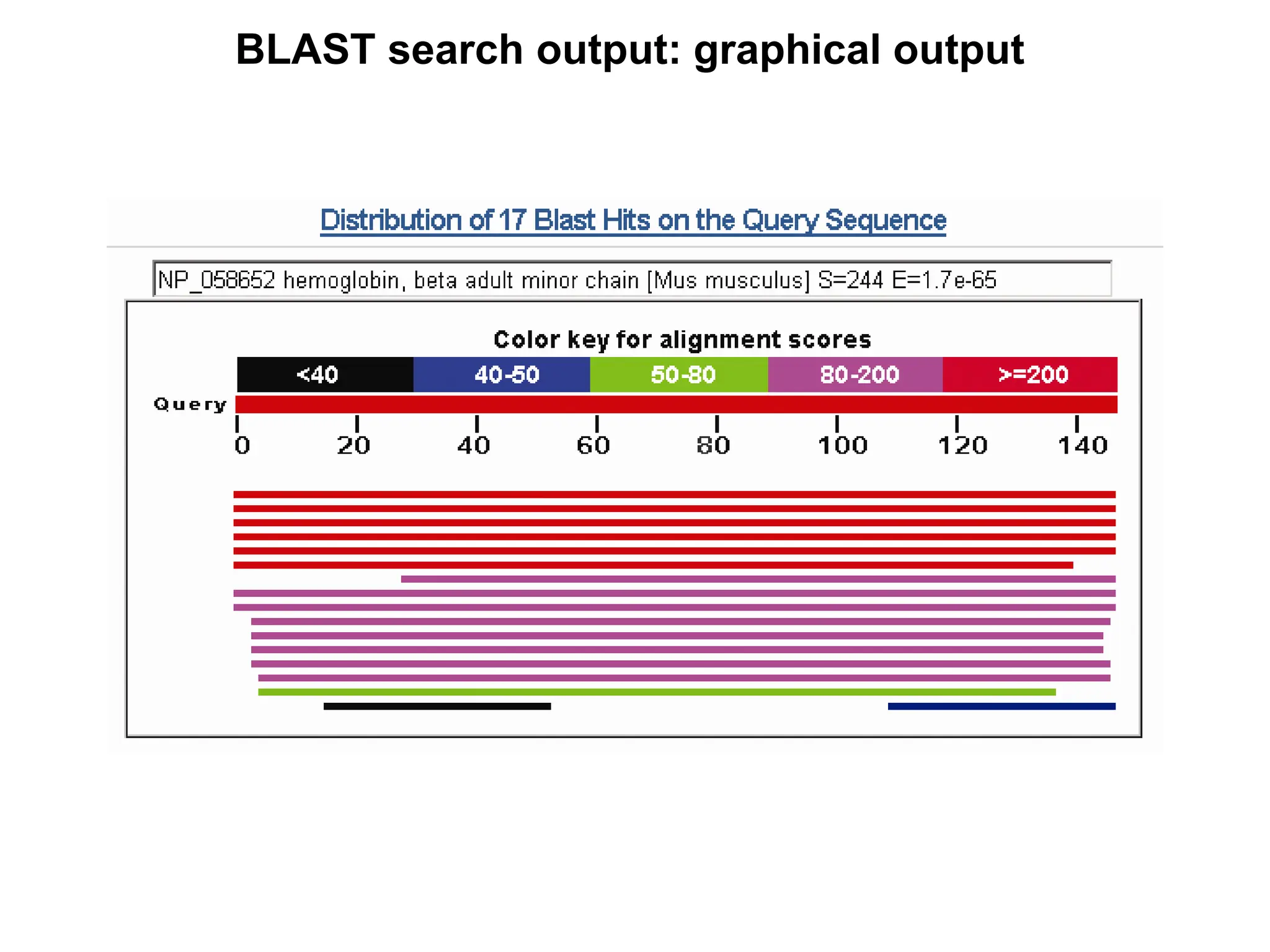
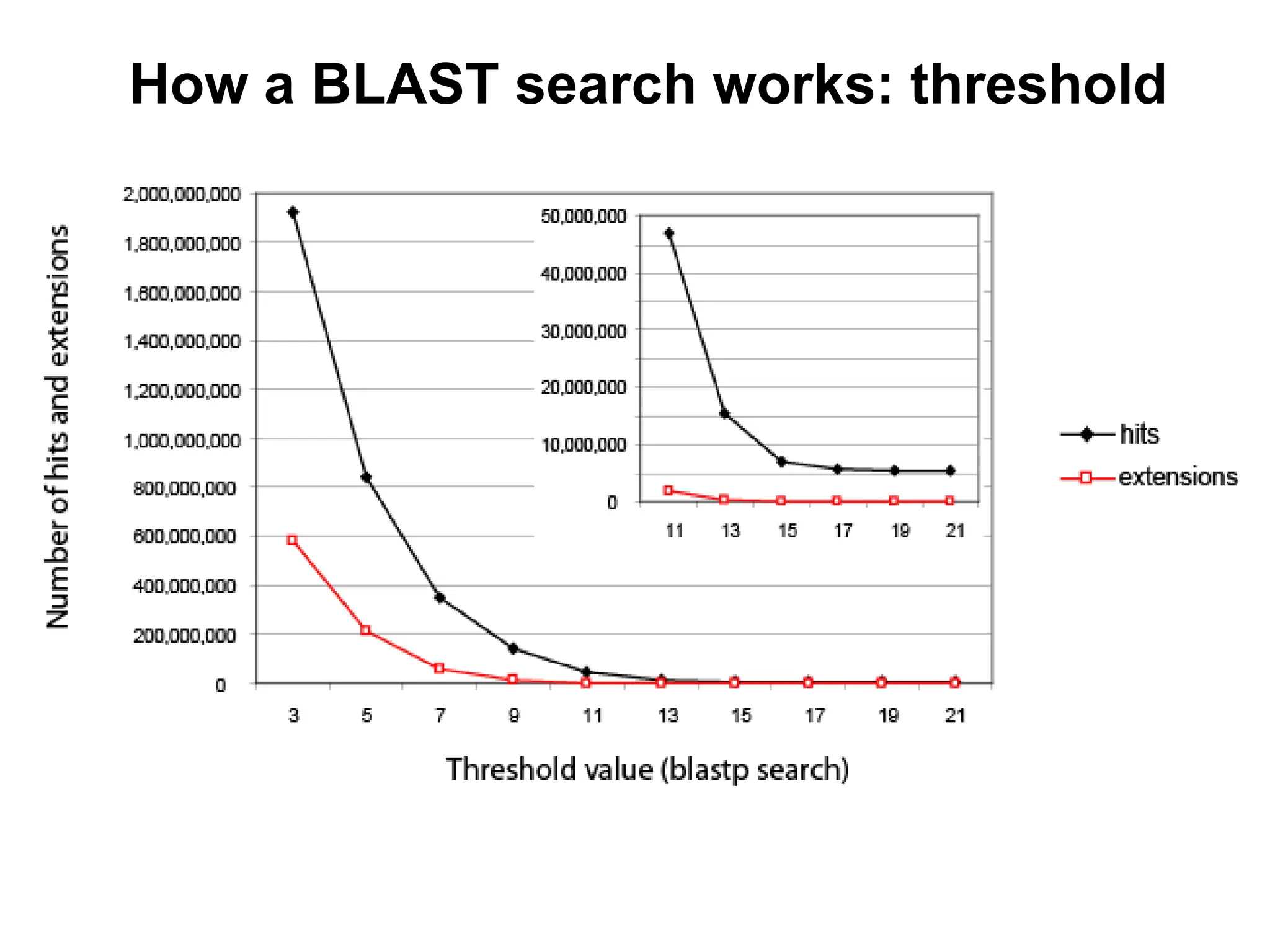
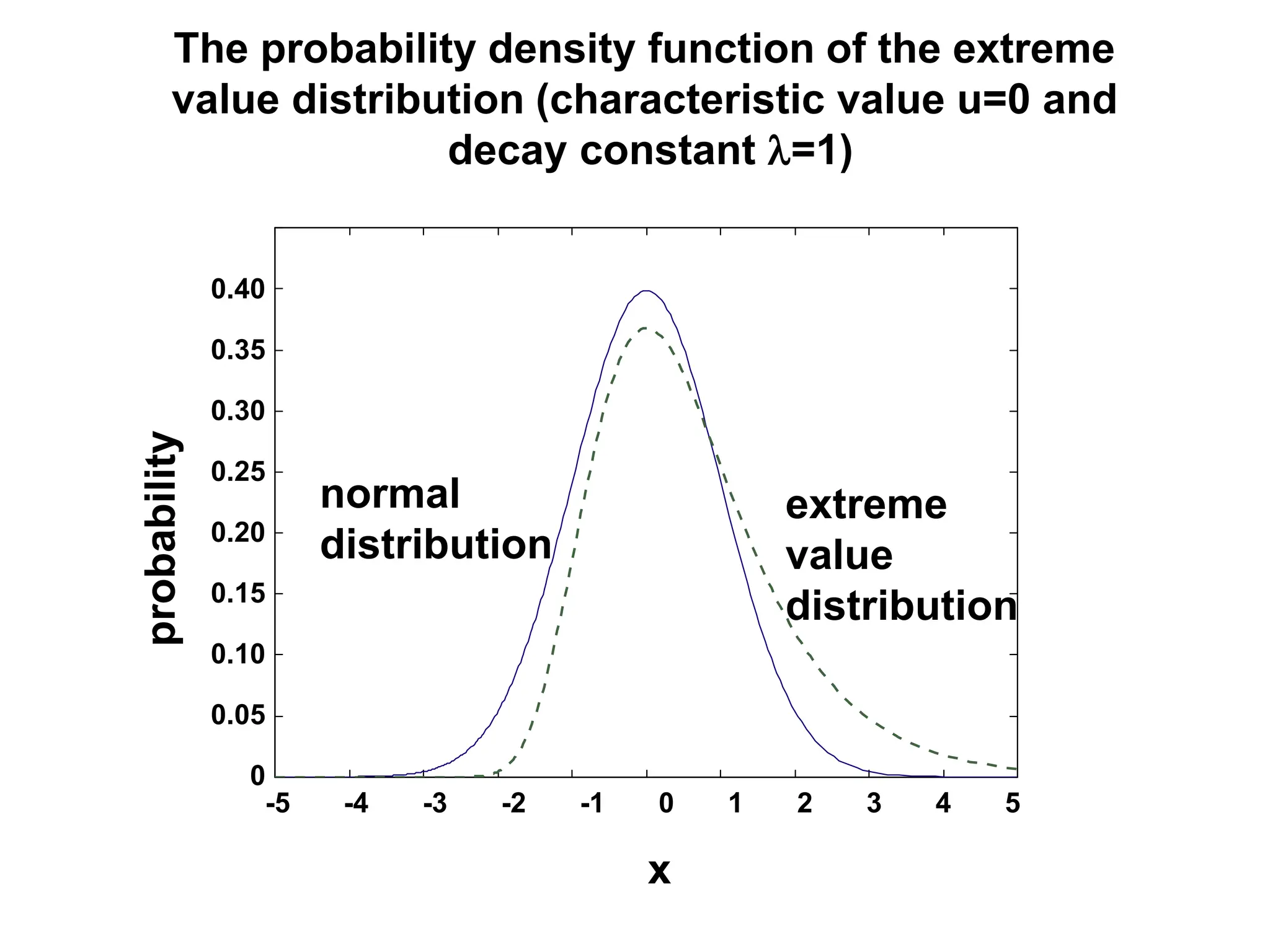
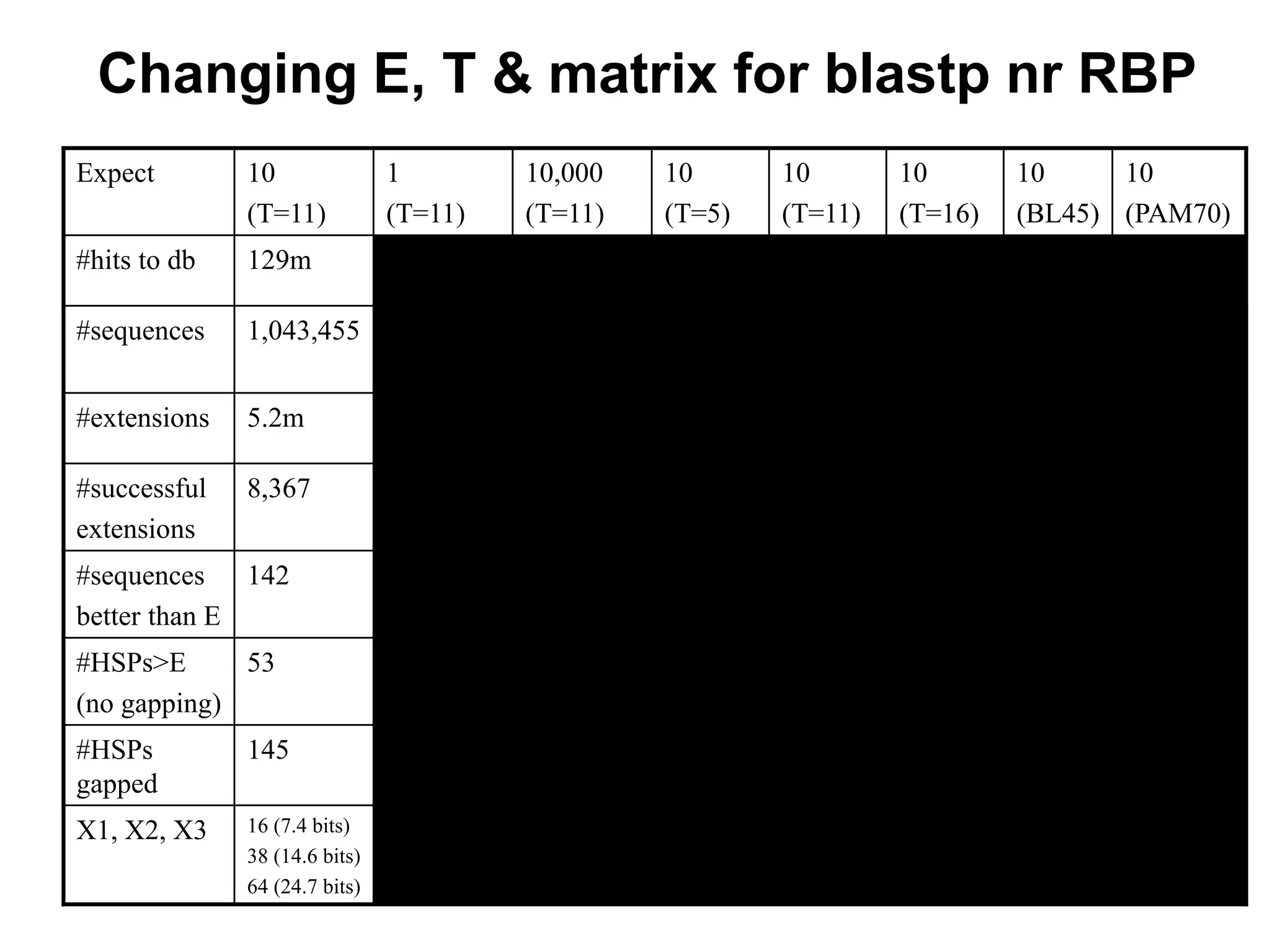
This document discusses the BLAST algorithm for comparing biological sequences. It explains that BLAST allows rapid sequence comparison of a query sequence against a database. BLAST is fast, accurate, and accessible online. The document then describes the four main components of a BLAST search: choosing the query sequence, BLAST program, database, and optional parameters. It provides details on how to interpret BLAST search results, including the expect value, and how BLAST works by compiling word pairs from the query and database in three phases of searching and alignment.