Download as PDF, PPTX


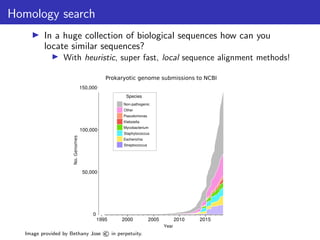




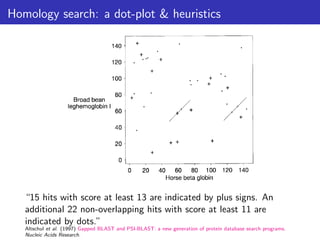
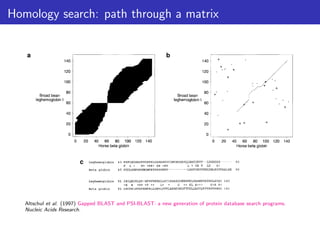

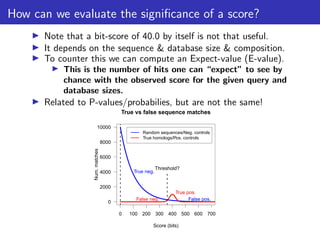
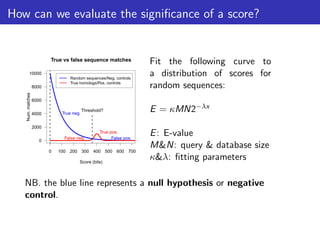





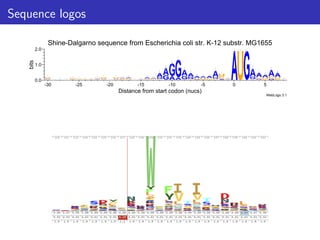
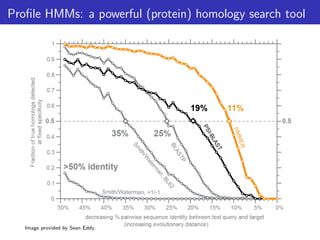



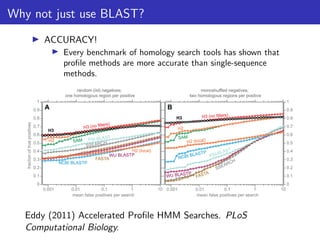
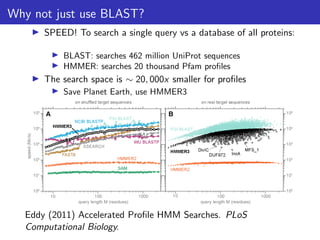










The document discusses homology search and comparative genomics, focusing on tools like BLAST and Profile HMMs for identifying similar biological sequences. It explains the functioning of BLAST, the significance of E-values, and compares the accuracy and applications of different homology search methods. It emphasizes the importance of proper evaluation when interpreting sequence similarity results and highlights that Profile HMMs generally provide better accuracy for divergent sequences compared to BLAST.



































































